Embed Size (px)
Citation preview

Les Puces à ADN sur lames de verre : principes et méthodes de confection, d’application
expérimentale et d’analyse des données.
Waka LIN Extraits du mémoire de la thèse de doctorat : « Applications de la technologie des Puces à ADN à l’étude de la différenciation méiotique et des mécanismes de recombinaison chez la levure Saccharomyces cerevisiae » Soutenue le 27 avril 2004. Equipe Alain NICOLAS UMR144 CNRS – Institut Curie, Section de Recherche

1
Sommaire
I. Introduction : la technologie des puces à ADN.............................................................5 1. Avancées de la génomique fonctionnelle chez la levure.............................................5 2. Définition et principaux types de puces à ADN..........................................................6 3. Application à l’analyse de l’expression transcriptionnelle..........................................8
3.1. Principe général .......................................................................................................................... 8 3.2. Les études comparatives de transcriptomes .............................................................................. 10 3.3. Autres variantes de l’analyse de l’expression transcriptionnelle .............................................. 11
4. Application à l’analyse des génomes ........................................................................11 4.1. La localisation des sites d’interaction de protéines avec la chromatine.................................... 12 4.2. La détection du nombre de copies d’ADN par CGH-array....................................................... 14 4.3. La détection de mutations et le génotypage de polymorphismes.............................................. 15 4.4. La détection et le criblage d’organismes spécifiques et de souches portant des marqueurs
d’identification.......................................................................................................................... 15
II. Confection des puces de type « spotted microarrays »...............................................17 1. Amplification des sondes par PCR............................................................................17
1.1. Choix des produits à déposer .................................................................................................... 17 1.2. Amplification par PCR, purification et stockage des sondes d’ADN ....................................... 18 1.3. Contrôle des produits de PCR par électrophorèse..................................................................... 19
2. Impression robotisée des lames .................................................................................20 2.1. Types de lames utilisées pour l’impression .............................................................................. 20 2.2. Dépôt des produits de PCR par le robot.................................................................................... 21 2.3. Coordonnées des gènes sur la matrice imprimée ...................................................................... 22
3. Traitement de finition et préhybridation des lames ...................................................23
III. Préparation des cibles et hybridation des puces .........................................................24 1. Préparation des échantillons d’ARN .........................................................................24
1.1. Extraction des ARN totaux et messagers.................................................................................. 24 1.2. Mesure de la concentration et contrôle de la qualité des ARN ................................................. 25
2. Transcription inverse et incorporation des marqueurs fluorescents ..........................27 3. Hybridation et lavage.................................................................................................28
IV. Acquisition et analyse des données ..............................................................................29 1. Acquisition des données brutes .................................................................................29
1.1. Lecture sur le scanner ............................................................................................................... 29 1.2. Extraction des données numériques.......................................................................................... 30
2. Transformation et stockage des données ...................................................................31 2.1. Filtrage primaire des données brutes ........................................................................................ 31 2.2. Le calcul des ratios d’expression .............................................................................................. 31 2.3. La normalisation ....................................................................................................................... 32 2.4. Le stockage dans des bases de données et le suivi des expériences.......................................... 34
3. Analyse et interprétation des résultats .......................................................................36 3.1. La mesure de l’expression différentielle ................................................................................... 36 3.2. Le regroupement hiérarchique des profils d’expression ........................................................... 38 3.3. La comparaison avec les données existantes ............................................................................ 41 3.4. Réflexions sur la conception de l’expérience biologique.......................................................... 43

2
V. Guide de diagnostic des anomalies...............................................................................46 1.1. Problèmes liés à l’aspect des spots ........................................................................................... 47 1.2. Problèmes de bruits de fond sur la lame ................................................................................... 48 1.3. Faible intensité de signal........................................................................................................... 49 1.4. Données biaisées détectées à l’analyse ..................................................................................... 50
VI. Protocoles expérimentaux.............................................................................................51
VII. Références bibliographiques.........................................................................................56

3
Liste des abréviations : ADN Acide DésoxyriboNucléique ADNc ADN complémentaire aa-dUTP amino-allyl dUTP AMAD Another MicroArray Database APC Anaphase-Promoting Complex APS Ammonium Persulfate ARN Acide RiboNucléique ARNm ARN messager ARNr ARN ribosomique ATM Ataxia-Telangiectasia Mutated ATR ATM-Rad3-Related BASE BioArray Software Environment BSA Bovine Serum Albumin CDBs Cassures Double-Brin CEA Commissariat à l’Energie Atomique CGH Comparative Genomic Hybridization Cdk Cyclin-dependent kinase ChIP Chromatin Immuno-Precipitation Cy3 Cyanine 3 Cy5 Cyanine 5 DAPI Diamino-Phenylindole dATP désoxy Adénosine Triphosphate dCTP désoxy Cytosine Triphosphate DEPC Diéthyl Pyrocarbonate dGTP désoxy Guanine Triphosphate DMSO Diméthyl Sulfoxyde DNase Désoxyribonucléase dNTP désoxy Nucléotide Triphosphate dUTP désoxy Uracile Triphosphate DTT DithioThréitol dTTP désoxy Thiamine Triphosphate EDTA Acide Ethylène Diamine Tétra-acétique ENS Ecole Normale Supérieure ESPCI Ecole Supérieure de Physique et
Chimie Industrielle FACS Fluorescence-Activated Cell Sorting GO Gene Ontology HEPES Hydroxyethyl Piperazine Ethanesulfonic HU Hydroxyurée Ir Intensité du signal rouge Iv Intensité du signal vert
kb kilo paires de bases kDa kilo Dalton Lowess Locally weighted scatter plot smoothing MI 1re division de méiose MII 2e division de méiose MAGEML MicroArray Gene Expression Markup
Language MGED Microarray Gene Expression Database MIAME Minimal Information About a Microarray
Experiment MIPS Munich Information center for Protein
Sequences MMS Méthyl Méthane Sulfonate MSE Middle Sporulation Element NHS-ester N-Hydroxysuccinimidyl ester NRE Negative Regulatory Element ORF Open-Reading Frame pb paire de bases PCR Polymerase Chain Reaction PPG Polypropylène Glycol RNase Ribonucléase RNR Ribonucléotide Réductase Rr/v Ratio des signaux d’hybridation
rouge/vert Rt/0h Ratio d’expression au temps t, relatif au
temps initial t=0h SAGE Serial Analysis of Gene Expression SDS Sodium Dodécyl Sulfate SGD Saccharomyces Genome Database SNP Single-Nucleotide Polymorphism SSC Saline Sodium Citrate TCA Trichloro Acetic Acid TE TRIS-EDTA TEMED Tétraméthyl Ethylène Diamide UV Ultra-Violet UAS Upstream Activator Sequence URS1 Upstream Repressor Sequence 1 yMGV yeast Microarray Global Viewer YPD Yeast Proteome Database, ou milieu
Yeast Peptone Dextrose

4
Avertissements – nomenclature utilisée dans ce document :
- Dans le texte qui suit, les fragments d’ADN fixés à la surface de la puce sont appelés « sondes » (« probe » en anglais), et les séquences nucléiques contenues dans l’échantillon à analyser sont appelées « cibles » (« target » en anglais), comme il a été convenu et recommandé notamment dans le manuel « DNA Microarrays » (Bowtell et Sambrook, 2003) [3], et le dossier « The Chipping Forecast II » (Nature Genetics supplement, 2002) [4]. Les termes sont parfois inversés selon les publications.
- Pour faciliter la lecture, j’ai conservé certains termes spécialisés couramment utilisés en anglais sans les traduire systématiquement en français, notamment « spot » (dépôt de fragments d’ADN sur une puce à ADN), « ORF » (open-reading frame = cadre ouvert de lecture), « checkpoint » (mécanisme de surveillance moléculaire induisant une réponse cellulaire aux dommages à l’ADN, aux défauts de réplication ou de recombinaison), « cluster » (ensembles de données obtenus grâce à une analyse par regroupement hiérarchique), « ChIP-on-Chip » (chromatine-immunoprécipitation et analyse sur puces à ADN).
- En accord avec la nomenclature internationale, les noms des gènes de S. cerevisiae sont indiqués en italiques, en majuscule lorsque le gène est sauvage (exemple : SPO11) et en minuscule lorsqu’il est inactivé et récessif (spo11). Les protéines correspondantes sont désignées avec une lettre initiale majuscule (Spo11).

5
I. Introduction : la technologie des puces à ADN
1. Avancées de la génomique fonctionnelle chez la levure
L’essor fulgurant des études de génomique fonctionnelle, faisant suite aux avancées des projets internationaux de séquençage des génomes amorcés à la fin des années 80, suscite aujourd’hui un très grand intérêt expérimental et conceptuel ([5] pour revue). Les séquences des génomes entiers de nombreux organismes sont désormais connues – plus de 100 bactéries, 1000 virus, et plusieurs eucaryotes modèles tels que les levures S. cerevisiae, S. pombe, C. albicans, la drosophile D. melanogaster, l’arabidopsis A. thaliana et le nématode C. elegans1 [6] – ainsi que de la majeure partie du génome humain publié en avril 2003 [7, 8]. L’enjeu réside à présent dans la caractérisation de la fonction des gènes et des voies de régulation leur permettant de réaliser les processus biologiques dans lesquels ils sont impliqués. Des outils et des stratégies innovants se sont développés afin de répondre aux besoins d’une exploration par des analyses fonctionnelles systématiques à l’échelle de la globalité du génome. La levure boulangère Saccharomyces cerevisiae est reconnue depuis longtemps comme un modèle de choix pour les études génétiques classiques. Ce microorganisme est non pathogène, a une croissance rapide dans des conditions de cultures simples, en étant stable aussi bien a l’état haploïde que diploïde, et la grande efficacité de la transformation par recombinaison homologue facilite l’introduction de mutations et l’application de nombreuses techniques d’analyses moléculaires et cellulaires. S. cerevisiae se prête aussi particulièrement bien au développement des méthodes d’analyse génomique à haut débit. Elle a été le premier organisme eucaryote dont le génome a été séquencé, grâce à un programme de collaboration européen puis mondial initié par A. Goffeau en 1989 et achevé en 1996 [9]. Son génome est relativement petit, comportant un total d’environ 12 mégabases réparties sur 16 chromosomes. Les bases de données de référence (SGD2 [10], MIPS3 [11], YPD4 [12]) recensent aujourd’hui environ 5800 ORFs confirmés et plus de 800 ORFs dits « hypothétiques ». Près de 70% de l’ADN non-ribosomal code pour des protéines et très peu de gènes (263) contiennent des introns. Enfin, la plupart des fonctions cellulaires fondamentales caractérisées chez S. cerevisiae sont conservées chez les organismes supérieurs : près de 50% des gènes humains impliqués dans des maladies génétiques héréditaires ont des homologues identifiés chez la levure. La connaissance de son génome offre ainsi un grand intérêt aussi bien pour l’étude de l’évolution et de l’adaptation des espèces que pour prédire la fonction de gènes chez l’homme.
1 Site d’accès à GenBank par Entrez au NCBI : http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db = Genome 2 Site de Saccharomyces Genome Database : http://www.yeastgenome.org/ 3 Site du Munich Information Center for Protein Sequences : http://mips.gsf.de/genre/proj/yeast/index.jsp 4 Site du Proteome BioKnowledge Library de Incyte : https://www.incyte.com/control/tools/proteome

6
Parmi les gènes identifiés chez S. cerevisiae, plus de 2000 codent pour des protéines de fonction inconnue [9, 13]. L’attention portée à l’étude fonctionnelle chez la levure a conduit à l’émergence de nombreuses approches d’analyses globales ([14, 15] pour revue) : production et caractérisation phénotypique de collections de mutants (issus de mutagenèse aléatoire [16], de délétion systématique de chaque ORF du génome [17, 18] ou d’insertion de marqueurs protéiques [19, 20]), analyse de l’expression transcriptionnelle [21, 22] ou protéique [23] sur « biopuces », études d’interactions protéines-protéines par des systèmes « double-hybride » à grande échelle [24, 25], d’interactions protéines-chromatine par « ChIP-on-Chip » [26, 27] et études protéomiques par analyses biochimiques [28] ou spectrométrie de masse [29, 30]. Dans ce contexte, la technologie des « puces à ADN » s’est imposée en quelques années comme un outil majeur de la génomique fonctionnelle.
2. Définition et principaux types de puces à ADN
Une puce à ADN, aujourd’hui communément appelée « DNA microarray » en anglais (de « array » = rang ordonné), est constituée de fragments d’ADN immobilisés sur un support solide selon une disposition ordonnée. Son fonctionnement repose sur le même principe que des technologies telles que le Southern blot ou le northern blot, qui sont couramment utilisées pour détecter et quantifier la présence d’une séquence nucléique spécifique au sein d’un échantillon biologique complexe, par hybridation à une sonde de séquence complémentaire portant un marquage radioactif [31]. La confection des puces à ADN a permis d’étendre ce principe à la détection simultanée de milliers de séquences en parallèle. Une puce comporte quelques centaines à plusieurs dizaines de milliers d’unités d’hybridation appelées « spots » (de l’anglais spot=tache), chacune étant constituée d’un dépôt de fragments d’ADN ou d’oligonucléotides correspondant à des sondes de séquences données. L’hybridation de la puce avec un échantillon biologique, marqué par un radioélément ou par une molécule fluorescente, permet de détecter et de quantifier l’ensemble des cibles qu’il contient en une seule expérience. D’abord conçues sur des membranes poreuses de nylon (appelées parfois « macroarrays » par opposition aux « microarrays »), les puces à ADN ont été progressivement mises au point sur lames de verre à la fin des années 90. La miniaturisation, rendue possible par l’utilisation d’un support solide, de marqueurs fluorescents et par les progrès de la robotique, permet aujourd’hui de fabriquer des puces comportant une très haute densité de spots, susceptibles de recouvrir l’intégralité du génome d’un organisme sur une simple lame de microscope. On distingue plusieurs types de puces selon la densité des spots, le mode de fabrication, la nature des fragments fixés à la surface et les méthodes d’hybridation. Les caractéristiques des puces les plus courantes sont résumées dans le Tableau 1.

7
« Macroarray »
- support : membrane de nylon
- taille des spots : 0,5-1mm
- densité : quelques centaines de spots/cm2
- sondes : produits de PCR
- cibles : ADNc avec marquage radioactif au 32P
- principales applications : analyse de l’expression des gènes
« Microarray spottée »
- support : lame de verre à revêtement chimique
- taille des spots : ~100µm
- densité : 1000-10000 spots/cm2
- sondes : produits de PCR ou oligonucléotides longs (30-70mers)
- cibles : ADNc ou produits de PCR avec marquage fluorescent au Cy3 et Cy5
- principales applications : analyse de l’expression, ChIP-on-Chip, CGH-array
« GeneChips » de Affymetrix
- support : lame de verre à revêtement chimique
- taille des spots : ~20µm
- densité : jusque 250000 spots/cm2
- sondes : oligonucléotides courts (20-25 mers) synthétisés in situ
- cibles : ARNc ou produits de PCR avec marquage fluorescent à la biotine-streptavidine
- principales applications : analyse de l’expression, détection de marqueurs moléculaires
Tableau 1 : Principaux types de puces à ADN
Les deux technologies dominantes sont les puces dites « spottées » par un dépôt robotisé de produits de PCR ou de longs fragments oligonucléiques (« spotted microarrays ») et les puces à oligonucléotides synthétisés in situ :
La méthode de fabrication des puces « spottées » a été développée par l’équipe de P. Brown à l’Université de Stanford, aux Etats-Unis [21]. Elle est aujourd’hui bien établie et de nombreuses plate-formes de production sont implantées dans les laboratoires académiques. Des solutions d’ADN sont préparées soit par amplification PCR à partir du génome ou de banques d’ADN complémentaires, soit par synthèse d’oligonucléotides longs (30-70 mers). Des micro-gouttelettes de ces solutions sont ensuite déposées par un robot, selon une matrice d’emplacements définis, sur une lame de verre traitée par un revêtement chimique qui permet de fixer l’ADN. En général, chaque spot de la matrice correspond à un gène donné. Les robots nécessaires à la fabrication de ces puces étaient construits à l’origine de manière artisanale dans chaque laboratoire selon le modèle conçu par J. DeRisi et dont les plans de montage et le logiciel de pilotage sont disponibles sur Internet 5 . Aujourd’hui, il existe plusieurs modèles commerciaux
5 Site The MGuide. Version 2.0 : http://cmgm.stanford.edu/pbrown/mguide/index.html

8
permettant d’obtenir des rendements de quelques dizaines à plusieurs centaines de lames en une seule série de production de quelques heures. Nous utilisons actuellement ce type de puces au laboratoire et je détaillerai les méthodes de fabrication et d’utilisation dans la suite de ce document.
Les puces à oligonucléotides synthétisés in situ par photolithographie [32]
(« GeneChips » de la société Affymetrix) ou par impression « jet d’encre » [33] (Agilent Technologies / Rosetta Inpharmaceutics) ne peuvent être produites que par des sociétés industrielles spécialisées, mais elles sont également de plus en plus utilisées et elles bénéficient désormais d’une importante diversification, d’une certaine baisse des prix et d’un contrôle de qualité accru. Une contrainte souvent posée par l’utilisation de ces puces est qu’elle nécessite en général l’emploi de méthodes et d’équipements imposés par le fournisseur (type de lecteurs, de logiciels d’analyse…) et que les licences de propriété industrielle ne permettent pas l’accès à certaines informations (telles que la séquence des sondes présentes sur la puce). La dépendance vis-à-vis de ces sociétés commerciales reste ainsi très forte. Il a été aussi souvent reproché un certain manque de souplesse, comme notamment les contraintes de production ne permettaient pas de fabriquer en quantité réduite des puces spécifiques à des besoins particuliers, mais d’importants progrès semblent avoir été accomplis à ce niveau face à l’accroissement de la demande (fabrication de puces à façon, diversification et adaptation des types de puces proposés à de nouvelles applications et de nouveaux organismes…).
Quel que soit le type de puces, le succès de la technologie a entraîné, depuis le début des années 2000, un élargissement considérable du choix des équipements et des protocoles expérimentaux, aussi bien pour la fabrication des lames que pour l’amélioration des conditions de manipulation en vue d’optimiser la sensibilité, la spécificité et la reproductibilité de la méthode ([34] pour revue). Les études exploitant l’utilisation des puces à ADN se multiplient rapidement dans des domaines d’application variés. Désormais, l’accent est mis prioritairement sur le besoin de rationaliser la gestion des expériences et de développer des systèmes de stockage et d’analyse de la masse de données générée. Les paragraphes suivants présentent les applications les plus courantes, illustrées d’exemples choisies en particulier parmi les nombreuses études publiées sur la levure S. cerevisiae.
3. Application à l’analyse de l’expression transcriptionnelle
3.1. Principe général
L’analyse de l’expression transcriptionnelle des gènes (parfois appelée « expression profiling ») a été la première application pour laquelle la technologie des puces à ADN a été développée avec succès. C’est encore l’application largement dominante aujourd’hui. L’hybridation d’un échantillon biologique sur une puce à ADN permet d’identifier et de

9
mesurer l’abondance des espèces d’ARN messagers (ARNm) qu’il contient. Dans la suite de mon étude, pour simplifier, le terme expression désignera, sauf indication contraire, l’expression transcriptionnelle, c’est-à-dire la production d’ARNm transcrits à partir de la matrice d’ADN d’un gène activé. L’utilisation des puces « spottées » permet d’acquérir une mesure relative du niveau
d’expression des gènes dans un échantillon cellulaire par rapport à un témoin de référence, par exemple une souche mutée comparée à une souche sauvage, ou des cellules cultivées dans deux conditions différentes. Le principe expérimental est résumé dans la Figure 1:
Cibles d’ADNc avec marquage fluorescent
ADN à déposer
Impression robotisée
référenceéchantillon
Extraction d’ARNm
Cy3Cy5
Hybridation et lecture
a
b
c
Figure 1 : Principe général de l’analyse de l’expression transcriptionnelle sur une puce de type « spottée »
(a) L’ensemble des ARNm sont extraits des prélèvements de cultures cellulaires à analyser.
(b) Une transcription reverse est réalisée en présence de nucléotides modifiés permettant de coupler un marqueur fluorescent. Des cibles d'ADN complémentaires (ADNc) représentatives de l'ensemble des gènes exprimés pour chaque culture sont ainsi obtenues.
(c) Les deux échantillons sont marqués par des fluorochromes à spectres d’émission distincts (les plus couramment utilisées sont les carbocyanines Cy3 et Cy5) et hybridées simultanément sur une même puce. Au contact de la puce, les brins d'ADNc marqués s'apparient avec les sondes de séquence complémentaire sur la lame. La mesure de l’intensité du signal fluorescent émis sur chaque spot permet ainsi d’estimer le taux d’expression différentiel du gène correspondant.
L’utilisation de puces à oligonucléotides de type Affymetrix permet de quantifier en
théorie l’abondance absolue de chaque ARNm transcrit. Les ARNm de l’échantillon à

10
analyser sont amplifiés, fragmentés et marqués par un système de couplage biotine-streptavidine pour l’hybridation sur la puce. Chaque gène est représenté sur la puce par une quinzaine de sondes constituées d’oligonucléotides courts de 20-25 bases couvrant différentes portions spécifiques du gène. Une estimation directe du niveau d’expression de chaque gène est obtenue en calculant le signal moyen sur l’ensemble des sondes représentant le gène.
3.2. Les études comparatives de transcriptomes
Le transcriptome est la population des ARNm exprimés par un organisme à un instant donné. Il résulte d’un équilibre entre la synthèse et la dégradation des ARNm et varie en fonction des conditions intra- et extra-cellulaires. Il offre ainsi une représentation dynamique de l’état de la cellule et des processus biologiques en cours. L’analyse du transcriptome permet d’établir le « profil d’expression » de chaque gène considéré, c’est-à-dire la variation de son niveau d’expression selon un ou plusieurs paramètres (temps, type cellulaire, etc.). De nombreuses études ont ainsi été réalisées dans différents organismes afin d’identifier les gènes co-régulés dans certaines réponses cellulaires spécifiques. Chez la levure, nous pouvons citer notamment les études de la variation transcriptionnelle des gènes :
- au cours du cycle cellulaire mitotique [35, 36], - au cours de la méiose et de la sporulation [37, 38], - en réponse à la transition de la fermentation anaérobie à la respiration [21, 39], - en réponse aux lésions de l’ADN provoquées par des irradiations ou des agents
génotoxiques [40], etc. Plus de 80 publications présentant des résultats d’expériences de puces à ADN réalisées chez la levure sont actuellement recensées dans la base de données yMGV6. La comparaison de divers transcriptomes de mutants, de types cellulaires ou de tissus donnés permet également de prédire la fonction de gènes non caractérisés et d’élucider des réseaux de régulation de voies biochimiques complexes. Hughes et al. ont ainsi démontré qu’il est possible d’assigner de nouvelles fonctions à des gènes de levure en comparant les transcriptomes de mutants portant une délétion de ces gènes à un ensemble de 300 profils d’expression générés dans une étude systématique [41]. Enfin, l’analyse du transcriptome permettant de caractériser l’état de la cellule, l’utilisation de puces à ADN dans un objectif de diagnostic est en développement. En particulier, cette approche permet de distinguer des types de cancers non différentiables par d’autres méthodes et ouvre des perspectives de classification de tumeurs [42, 43]. Elle est aussi très utilisée pour la recherche et la validation de substances thérapeutiques en permettant l’identification de nouveaux gènes-cibles et la caractérisation de la réponse cellulaire à un traitement. 6 Site du yeast Microarray Global Viewer : http://www.transcriptome.ens.fr/ymgv/

11
3.3. Autres variantes de l’analyse de l’expression transcriptionnelle
Outre les analyses des variations globales du transcriptome d’un organisme, des études plus récentes ont permis d’étendre le principe de la mesure de l’expression transcriptionnelle vers d’autres types d’applications spécifiques, en majorité développées chez la levure. Notamment, les puces à ADN ont été utilisées pour caractériser :
- le contrôle de la dégradation des ARNm après arrêt de la transcription [44], - la nature des ARNm associés aux polysomes membranaires [45] ou mitochondriaux
[46] par séparation fractionnée des compartiments cellulaires et comparaison avec les ARNm cytosoliques,
- la spécificité de la répression transcriptionnelle par siRNA (small interfering RNA) [47, 48],
- l’association des ARNm aux ribosomes pour mesurer le taux de traduction des protéines [49].
La confection de puces spécialisées, contenant des spots correspondants aux ARN non-codants, permet aussi d’analyser :
- le taux d’épissage des ARNm dans différents mutants, les puces utilisées portant des sondes oligonucléiques correspondant à chaque exon, intron, et à la jonction exon/intron, ce qui permet de différencier les ARNm épissés de ceux contenant encore un intron [50].
- le taux de production et de maturation des ARN non-codants, les puces portant des sondes correspondant aux transcrits primaires, aux fragments épissés et aux produits finaux de différents ARN non-codants : ARNr, ARNt, ARN du splicéosome et petits ARN nucléolaires [51].
Cette approche a été également utilisée pour détecter l’expression des exons et des variants d’épissage chez l’homme afin de réaliser une première ébauche d’annotation du génome humain [48].
4. Application à l’analyse des génomes
D’autres applications des puces à ADN, développées plus récemment, visent à caractériser des propriétés du génome au sein de la cellule telles que sa structure physique, ses interactions avec des protéines régulatrices, et les modifications épigénétiques qu’il peut subir. Ces approches permettent d’élucider le rôle de la structure dynamique du génome dans la régulation de fonctions cellulaires fondamentales comme la transcription, la réplication, la recombinaison, la ségrégation chromosomique et le maintien de la stabilité génomique.

12
4.1. La localisation des sites d’interaction de protéines avec la chromatine
La méthode de chromatine-immunoprécipitation a été développée à fin des années 90 afin d’identifier les sites d’interaction de protéines, telles que des facteurs de transcription, avec l’ADN génomique. Le principe est schématiquement représenté dans la Figure 2 :
Lysat cellulaire
Fragmentation
Incubation avec anticorps
Précipitation àl’agarose-protéine G
Lavage
PCR Semi-quantitative
Protéine àétudier
Hybridation sur puce après amplification et marquage
Identification des zones de fixation préférentielle le long des chromosomes
CENTEL TELCENTEL TEL
site chaud de fixation
-Ac +AcT
site froid
site chaud de fixation
-Ac +AcT
site froid
Autres protéines
ab
c
d
e
Figure 2 : Principe du « ChIP-on-Chip »
(a) Classiquement, les protéines sont fixées de manière covalente à l’ADN génomique par un
traitement au formaldéhyde (« cross-linking »).
(b) L’ADN est fragmenté par un traitement aux ultra-sons, et l’extrait cellulaire est incubé avec un anticorps spécifique de la protéine d’intérêt (ou spécifique d’un épitope avec laquelle la protéine aurait été marquée au préalable).
(c) La purification par immunoprécipitation permet d’isoler la protéine d’intérêt avec les fragments d’ADN qui lui sont associés.
(d) Une étude par PCR semi-quantitative permet de révéler, dans la fraction purifiée, un enrichissement en fragments contenant les séquences correspondant aux sites d’interaction.
(e) Le couplage de cette méthode à l’utilisation des puces à ADN permet de cartographier ces sites le long de l’ensemble des chromosomes. Les fragments d’ADN purifiés sont amplifiés, marqués à la fluorescence, et hybridés sur une puce simultanément à un témoin de référence. L’intensité du signal fluorescent sur un spot reflète alors la fréquence de fixation de la protéine au site correspondant.

13
Cette méthode, appelée souvent « ChIP-on-Chip » (Chromatin-ImmunoPrecipitation on Chip), a été surtout utilisée chez la levure, dont la taille restreinte du génome et la faible proportion des régions intergéniques facilite la construction de puces contenant ces régions, qui sont les cibles de facteurs de transcription. De nombreuses études ont été ainsi réalisées sur des facteurs connus tels que :
- les régulateurs du cycle cellulaire (complexes MBF, SBF, Mcm1, Swi5…) [27, 52], - l’activateur/répresseur de transcription Rap1 [53], - les activateurs de transcription Gal4 et Ste12 (induction de gènes en présence de
galactose et de phéromones) [26]. Une étude à grande échelle sur plus de 100 facteurs de transcription marqués d’un épitope Myc (sur 141 protéines décrites dans la base de données YPD7 comme ayant une activité de liaison à l’ADN et de transcription) a été aussi réalisée en 2002 par Lee et al. [54]. L’analyse de la distribution génomique des sites d’interaction des facteurs de transcription présente plusieurs intérêts :
Il permet de connaître directement l’ensemble des cibles naturelles de transcription lorsque leur identification par analyse de l’expression dans des souches mutées est impossible à réaliser (quand le facteur considéré est essentiel à la survie cellulaire et ne permet pas la construction de mutants), ou si elle est rendue difficile par l’influence des effets indirects ou pléiotropes induits par la mutation.
Il permet de caractériser la nature des mécanismes de reconnaissance de ces cibles : il est possible par exemple de déterminer la présence d’une séquence consensus commune à toutes les cibles ou de caractériser des domaines chromosomiques présentant une structure particulière (par exemple, les zones sub-télomériques réprimés par Rap1 et les inhibiteurs Sir qui lui sont associés [53]).
Enfin, il est possible d’analyser les mécanismes de recrutement de ces facteurs au sein des complexes protéiques associés à l’ADN. Une protéine peut interagir avec la chromatine non pas en se liant directement à l’ADN mais par l’intermédiaire d’autres facteurs. Nous pouvons ainsi hiérarchiser le recrutement des différents facteurs en déterminant quels sont ceux nécessaires à la fixation des autres.
Le principe du « ChIP-on-Chip » a été aussi étendu à la localisation d’autres types de sites spécifiques de la chromatine afin d’analyser de manière indirecte la dynamique de la structure du génome :
Deux approches différentes ont permis d’examiner les modifications structurelles des histones et les remaniements du nucléosome : la localisation des sites de fixation des protéines impliquées dans ces modifications (histone-déacétylase Rpd3 [55], histone-méthylase Set1 [56], complexe RSC de remaniement de la chromatine [57, 58]), et l’isolement direct des fragments correspondant aux sites modifiés (en effectuant une
7 Site du Proteome BioKnowledge Library de Incyte : https://www.incyte.com/control/tools/proteome

14
immunoprécipitation avec des anticorps spécifiques des résidus de lysine acétylés sur les histones [59]).
La méthylation de l’ADN, qui établit une répression de la transcription, peut être analysée de manière similaire en isolant les sites hyper-méthylés par digestion de l’ADN non modifié avec une enzyme de restriction sensible aux méthylations [60]. Ceci a permis par exemple de détecter des hyper-méthylations aberrantes au niveau des îlots CpG qui peuvent conduire au développement de cancers par répression de gènes suppresseurs de tumeurs.
Une étude a été également réalisée afin de caractériser les origines de réplication par localisation des sites de fixation des initiateurs de réplication ORC et MCM [61].
Enfin, Gerton et al. [62] ont établi la carte des sites de formation des cassures double-brin de l’ADN qui initient la recombinaison méiotique en isolant, par une méthode de filtration protéique [63], les fragments correspondant aux sites de coupure de la protéine Spo11, celle-ci restant liée de manière covalente à l’ADN dans certains mutants tels que rad50S. Dans notre laboratoire, nous avons pu également établir la carte des sites de formation des cassures double-brin méiotiques par une autre méthode : la chromatine-immunoprécipitation de la protéine Mre11 et de Spo11 marquée par un épitope HA [2].
4.2. La détection du nombre de copies d’ADN par CGH-array
La technique de la CGH (Comparative Genomic Hybridization [64]) permet de détecter les variations du nombre de copies d’ADN telles que les amplifications et les délétions de régions chromosomiques associées à des pathologies du développement ou à des cancers. En effet, dans une cellule cancéreuse, un gain en nombre de copies impliquerait une augmentation du taux d’expression d’oncogènes, tandis qu’une perte pourrait induire l’inactivation de gènes suppresseurs de tumeurs. Dans la CGH classique [64], les ADN génomiques d’un échantillon à tester (tissus tumoral) et d’un témoin de référence (tissus normal) sont extraits, marqués par des colorants fluorescents et hybridés sur un étalement de chromosomes métaphasiques, afin de permettre l’observation de remaniements chromosomiques. L’hybridation sur des puces à ADN représentant des portions connues du génome (appelées couramment « CGH-arrays ») permet de cartographier les réarrangements génomiques avec une résolution beaucoup plus élevée [65, 66]. Cette approche a été utilisée avec succès dans des études de classifications de sous-types de cancers, d’identification de gènes à expression amplifiée dans les tumeurs (cancers du sein, de l’estomac…) et d’analyse des réarrangements dans des syndromes génétiques constitutifs (anomalies congénitales…). Le principe de la CGH-array a pu être aussi appliqué dans des études fondamentales chez la levure telles que :

15
- l’analyse de la distribution et de la dynamique d’activation des origines de réplication, par différenciation et isolation de l’ADN répliqué ou non après incorporation d’isotopes lourds de carbone et d’azote [67],
- la caractérisation des réarrangements génomiques au cours de l’évolution de souches de levure soumises à une forte pression de sélection [68].
4.3. La détection de mutations et le génotypage de polymorphismes
Des puces à ADN à oligonucléotides courts, suffisamment spécifiques pour discriminer des séquences différant d’un seul nucléotide, sont utilisées afin de détecter des polymorphismes entre allèles d’un gène ou de nouvelles mutations. En particulier la détection des SNPs (« Single-Nucleotide Polymorphism ») présente un intérêt en oncologie et en pharmacologie en vue du diagnostic et du suivi des traitements (exemple [69]), par leur potentiel en tant que marqueurs génétiques stables permettant d’analyser les génotypes associés aux pathologies. La puissance de cette méthode pour identifier les variations alléliques et caractériser l’héritabilité des marqueurs a été illustrée chez la levure par Winzeler et al. [70]. Un ensemble de plus de 3700 marqueurs permettant de différentier deux souches de S. cerevisiae a été identifié par hybridation de leur génome sur des puces à oligonucléotides. Le suivi de la ségrégation de ces marqueurs dans la descendance du diploïde hétérozygote a permis de cartographier avec une haute résolution les évènements de recombinaison ayant eu lieu au cours de la méiose. De plus, l’établissement de la carte des marqueurs dans les ascendants a permis d’identifier les gènes responsables d’un caractère héréditaire simple, en comparant les génomes de plusieurs ségrégants présentant le même phénotype et en déterminant les régions chromosomiques communes.
4.4. La détection et le criblage d’organismes spécifiques et de souches portant des marqueurs d’identification
L’hybridation de cibles synthétisées à partir d’ADN génomique peut aussi permettre de détecter, au sein d’une population complexe, la présence d’un organisme dont on connaît un marqueur spécifique. Ainsi, les puces à ADN peuvent être utilisées pour détecter la présence de micro-organismes virulents ou caractéristiques d’une certaine condition de milieu (traitement des eaux, recherche de pathogènes…). Ce principe a été appliqué chez la levure pour réaliser des analyses phénotypiques systématiques sur la collection de mutants de délétion construite en 1999 par un consortium international [17]. Dans ce programme, pour chaque gène non-essentiel connu du génome de la levure (plus de 4200 gènes), une souche mutante a été construite en remplaçant le gène considéré par une cassette de délétion contenant une séquence oligonucléique unique (Figure 3a). Cette séquence, appelée « code-barres moléculaire », sert de marqueur d’identification

16
spécifique à chacune de ces souches. Une puce portant les séquences complémentaires à ces codes-barres permet alors d’analyser des mélanges complexes de différentes souches. Ceci a été appliqué à l’étude de l’évolution des mutants après croissance dans des conditions sélectives (Figure 3b). Les souches présentant une meilleure ou une moins bonne croissance dans la condition étudiée sont repérées dans le mélange par un signal plus ou moins fort émis par les spots correspondants de la puce. La représentativité relative des souches, reflétant un phénotype de croissance de chaque mutant, peut être ensuite interprétée par rapport à la fonction du gène inactivé.
KanMX
ORF de levure à remplacer
KanMX
Marqueurs « codes-barres » spécifiques de chaque
souche mutante
Amorces universelles permettant une
amplification par PCR
Transformation par recombinaison homologue
Mélange à densité égale de cellules de différentes souches
Culture en condition sélective
a b
Figure 3 : Principe de criblage d’un mélange complexe de souches : culture compétitive sur milieu sélectif dans le cas de la collection des mutants de délétion.
(a) Construction de souches mutées par remplacement d’un gène par un marqueur portant un « code-barres » d’identification.
(b) Test de croissance en milieu sélectif : la mutation confère un désavantage sélectif, donc le gène correspondant a un rôle potentiel pour favoriser la croissance dans les conditions étudiées. Le spot correspondant au « code-barres » de la souche ayant un défaut de croissance émet un signal plus faible sur la puce.
Cette méthode a été appliquée d’abord sur une partie de la collection de mutants (558 gènes) pour observer les défauts de croissance de certains mutants sur milieu minimum comparé au milieu riche [17], puis sur la collection complète pour analyser notamment :
- la croissance cellulaire dans différentes conditions de cultures sélectives [18], - la sensibilité aux irradiations UV et aux agents génotoxiques [71, 72], - la formation et la viabilité des spores en condition de culture méiotique [73], - les mutants déficients pour la réparation par « Non-Homologous End-Joining » [74], - les mutants synthétiques létaux avec les hélicases SGS1 et SRS2 [75].

17
II. Confection des puces de type « spotted microarrays »
1. Amplification des sondes par PCR
1.1. Choix des produits à déposer
Nous produisons au laboratoire des puces de type « spottées », selon la technologie développée par l’équipe de P. Brown à l’Université de Stanford. La première étape à réaliser est la préparation des fragments d’ADN qui constituent les sondes représentant chaque ORF ou région intergénique du génome étudié. Pour la levure, il existe aujourd’hui différentes sources de matériel pouvant être utilisées à cette fin :
Des produits d’amplification par PCR d’ORFs ou de régions intergéniques entiers :
Nous utilisons dans notre laboratoire cette méthode originellement employée chez P. Brown pour fabriquer les premières puces du génome complet de levure. Dans ce cas, les séquences correspondant à toute la longueur des ORFs et/ou des intergènes sont déposées sur la puce. Ces sondes de produits de PCR présentent notamment l’avantage de permettre aisément la réalisation d’expériences de type « ChIP-on-Chip ». Toutefois, les inconvénients sont qu’il est parfois difficile d’amplifier simultanément tous les fragments de tailles différentes et que les gènes présentant une forte homologie de séquence (plus de 70%) ne peuvent être distingués après hybridation. Des produits d’amplification par PCR d’une portion déterminée d’ORFs :
La société Research Genetics propose cette alternative en fournissant une banque d’amorces qui permet d’amplifier des fragments de 1 kb choisis sur chaque ORF de manière à faciliter l’amplification et à éviter si possible les régions conservées qui conduisnt à des hybridations croisées entre gènes homologues. Des oligonucléotides longs (30-70 mers) :
L’utilisation de banques d’oligonucléotides synthétisés à façon, de plus en plus développée aujourd’hui, permet de s’affranchir des problèmes d’hybridations croisées en choisissant les séquences des sondes dans les régions spécifiques non conservées de chaque gène. Elle permet aussi de réduire le coût et le temps de préparation par rapport à la fastidieuse étape d’amplification par PCR. La difficulté essentielle est de choisir les oligonucléotides assurant la meilleure spécificité et la meilleure sensibilité à l’hybridation, mais aujourd’hui plusieurs fournisseurs proposent des collections d’oligonucléotides « prêts à spotter » (Qiagen-Operon, MWG Biotech).

18
1.2. Amplification par PCR, purification et stockage des sondes d’ADN
Pour la synthèse des sondes, nous avons utilisé dans un premier temps une banque de matrices fournie par Research Genetics. Chaque matrice est constituée de la séquence entière d’un ORF de l’ATG au codon Stop, flanquée d’une séquence oligonucléique commune permettant une amplification par PCR de toutes les matrices avec une paire unique d’amorces universelles. Les deux premiers essais d’amplification des 6217 produits PCR de la banque (65 plaques à 96 puits au total) ont mobilisé le travail de quatre personnes pendant 3 mois chacun. Malheureusement, de nombreuses difficultés ont été rencontrées au niveau de l’efficacité de l’amplification, de la purification (différentes méthodes de purification ont été testées et conduisent à des concentrations et des qualités de produits différentes, cf. Guide de diagnostic des anomalies, p.46), et de la conservation des produits (les produits de certaines plaques se sont évaporés et/ou dégradés à cause d’un mauvais stockage dans le congélateur à –20 °C). Les puces que nous avions ainsi commencé à produire à partir du début de l’an 2000 étaient peu exploitables, car elles manquaient d’homogénéité au niveau de la forme des spots et de la concentration en ADN. Un troisième essai plus fructueux a été réalisé au cours d’un stage que j’ai effectué au laboratoire de J. DeRisi à l’University of California of San Francisco (UCSF), en octobre 2001. L’équipement du laboratoire DeRisi, mieux adapté à la production à grande échelle (16 thermocyclers pour PCR en plaques 96 puits, deux robots pipeteurs Beckmann, deux centrifugeuses Sorvall à plaques), a permis d’effectuer l’ensemble du travail en 3 semaines, deux personnes y participant. Les étapes sont résumées dans la Figure 4.
Amplification PCR de chaque ORF en plaque 96 puits
Vérification sur gel d’électrophorèse
Précipitation à l’isopropanolet séchage au speed-vac
Transfert en plaques 384 puits pour le dépôt
100 µl par puits, 65 plaques
5 µl par puits, 5 sets de 17 plaques
YAL001c
YAL002w
YPR204w
YAL001c
YAL002w
YPR204w
a
b
c
d
Amorces GenePairs
Figure 4 : Etapes de l’amplification des gènes-cibles par PCR.

19
(a) Une banque d’amorces spécifiques de chaque ORF (amorces Yeast GenePairs Primers de Research Genetics) a été utilisée pour amplifier à partir d’une matrice d’ADN génomique, ce qui a permis d’amplifier efficacement avec de meilleurs rendements.
(b) L’amplification est réalisée dans des plaques 96 puits grâce à une enzyme Taq polymérase (cf. Protocole 1, p.51). Les produits sont vérifiés par électrophorèse (cf. §II.1.3, p.19).
(c) Les produits sont purifiés par précipitation à l’isopropanol puis dissous dans un tampon 3x SSC et aliquotés dans des plaques 384 puits adaptées au dépôt robotisé. Nous avons choisi des plaques Genetix « pour microarrays » en polypropylène, permettant l’utilisation d’un très faible volume de solution (moins de 5 µl par puits) car les gouttes ne s’étalent pas au fond des puits comme dans des plaques en polystyrène.
(d) Les plaques sont stockées à –20 °C après lyophilisation dans un speed-vacuum, ce qui permet une conservation optimale et un bon contrôle du volume de liquide dans les puits. A chaque utilisation, les culots secs d’ADN sont re-dissous dans quelques microlitres d’eau stérile à 4 °C pendant 24h. Le volume d’eau utilisé est réduit à chaque série d’impression pour éviter une dilution des produits (5 µl la première fois, 4,75 µl ensuite etc.). On admet qu’une même plaque peut être ainsi réutilisée pour environ 10 séries d’impression successives.
1.3. Contrôle des produits de PCR par électrophorèse
Chaque produit de PCR a été vérifié par électrophorèse sur gel d’agarose 1%. La concentration optimale d’ADN pour l’impression des spots est d’environ 100 ng/µl. Cette concentration varie selon les gènes, mais nous avons pu montrer, lors de tests que j’ai conduits au laboratoire au cours de mon stage de DEA, que l’intensité du signal sur les spots diminue nettement s’ils sont déposés avec des concentrations 2 à 3 fois inférieures. Nous avons observé qu’une concentration trop élevée induit au contraire une augmentation du bruit de fond sur les lames à cause de la trop grande quantité de molécules non fixées à la surface. Les bandes correspondant à chaque produit de PCR sont visualisées et évaluées après coloration du gel par le bromure d’éthydium par comparaison à des marqueurs de masse moléculaire et nous avons annoté chaque produit selon qu’il soit :
0) « correctement » amplifié, 1) de faible concentration, 2) composé de deux bandes (dont une à la taille attendue), 3) composé de plusieurs bandes ou d’un « smear », 4) d’une taille différente de celle attendue, 5) absent.
Les plaques présentant plus de 10% d’échecs (produits notés 1-5) ont été ré-amplifiées. Nous éliminons systématiquement de l’analyse les spots correspondant aux produits notés 3-5.

20
Ceux notés 1 ou 2 sont conservés mais considérés avec prudence (un examen de la puce a posteriori a montré que beaucoup de produits notés 1 sont en réalité suffisamment amplifiés mais ont probablement été notés ainsi suite à un mauvais dépôt sur le gel d’agarose). Au final, nous avons obtenu 5812 / 6217 ORFs correctement amplifiés, soit un taux de réussite global de 93%.
2. Impression robotisée des lames
2.1. Types de lames utilisées pour l’impression
Le dépôt est effectué sur des lames en verre traitées par un revêtement chimique qui permet de fixer les brins d'ADN grâce à des interactions électrostatiques. Différentes types de lames sont disponibles aujourd’hui.
Des lames recouvertes de polylysine sont couramment utilisées, car elles sont simples et peu coûteuses à produire au laboratoire à partir de lames de microscope, par une série de trempages et de lavages successives dans une solution de PBS-polylysine. Cependant, nous avons rencontré de nombreuses difficultés car ces lames se conservent mal et sont souvent d’une qualité inégale d’une série à l’autre, selon le traitement et selon le lot de solution de polylysine commerciale utilisé (il semblerait que la qualité se soit détériorée depuis que le fournisseur Sigma-Aldrich a changé l’additif conservateur dans la formulation de leur solution de polylysine en 2002). Le revêtement de polylysine est fragile, il est souvent percé pendant l’impression, se déchire et parfois se détache de la surface de la lame pendant les traitements et lavages ultérieurs. Il est aussi peu homogène, et conduit à l’apparition de bruits de fond verts dans l’image dans les zones où il est irrégulier. Enfin, nous avons remarqué une dégradation survenant spontanément après quelques mois de stockage (parfois en à peine un mois) conduisant à une forte aggravation de ces défauts (cf. Guide de diagnostic des anomalies, p.46). Après plusieurs essais, nous avons aujourd’hui opté pour l’utilisation systématique
de lames commercialisées par Corning (lames CMT-GAPS II, à surface de gamma amino-propyl silane). Comme la polylysine, les charges positives des groupements amines permettent de fixer l’ADN. Ces lames sont coûteuses mais permettent d’obtenir en général d’excellents résultats. Elles sont plus résistantes aux variations de température, ne présentent pas de problème de déchirement et ont une longue durée de vie (2 ans selon Corning, mais leur utilisation a été encore possible après la date limite recommandée).

21
2.2. Dépôt des produits de PCR par le robot
Nous utilisons actuellement le robot OmniGrid II TAS de Biorobotics, installé en décembre 2001 à l’ENS dans une pièce à atmosphère contrôlée. Le robot, encore appelé « spotter » ou « arrayer », est muni de pointes métalliques fendues qui prélèvent par capillarité quelques nanolitres des produits d’une plaque de PCR et en déposent une micro-gouttelette par contact à la surface des lames (Figure 5). Un autre robot du constructeur Genemachines était utilisé avant cette date, mais nous en avons cessé l’emploi, étant donné la mauvaise qualité d’impression par rapport au nouveau système, due à une force de frappe mal adaptée et d’un mauvais contrôle des conditions d’humidité et de température.
x
Zy
1
2
3
4
a b c
Figure 5 : Le robot « spotter » de Biorobotics
(a) Vue d’ensemble. Le robot est équipé (1) d’un système de stockage et de distribution automatique de 24 plaques de PCR, (2) de quatre plateaux contenant 108 lames au total, (3) d’une tête d’impression contenant jusque 48 pointes, (4) d’un système de réglage de la température et de l’humidité. (b) Détail de la tête d’impression. Le plateau de lames se déplace selon x et le bras du robot selon y et z pour permettre le dépôt par contact. (c) Détail d’une pointe avec réservoir de solution à déposer. Diamètre réel : 100 µm.
Pour réaliser une puce comportant l’ensemble des ORFs du génome de la levure, nous effectuons un dépôt avec une tête d’impression équipée de 16 pointes (MicroSpot 2500) imprimant 16 spots simultanément. Nous déposons ainsi l’ensemble des spots (plus de 6200) avec un espacement de 290 µm sur une surface totale d’environ 2 cm x 4 cm, chaque spot mesurant 150-200 µm. Nous lançons des séries d’impression de 50 lames qui peuvent être réalisées en une nuit (17h). Un seul prélèvement des produits de PCR par les pointes permet d’imprimer sur plusieurs lames à la suite, mais nous nous sommes rendus compte que la quantité déposée diminue rapidement en fonction du nombre de lames, aussi nous programmons le robot de manière à ce qu’il prélève une nouvelle dose entre chaque plateau (toutes les 27 lames). La qualité de l’impression dépend de plusieurs paramètres tels que :
- le tampon de dissolution et la concentration en ADN des produits de PCR, - l’état des pointes (qui peuvent s’user ou se boucher), - l’humidité (maintenue à 50% pendant l’impression) et la température ambiante,

22
- la vitesse et la force de frappe des pointes sur les lames et dans les puits des plaques de PCR (le nouveau robot Biorobotics propose une option « soft touch » permettant de ralentir les pointes avant le contact),
- le nombre et la durée de lavages des pointes permettant d’éviter les contaminations inter-spots (3 cycles de 2 x 5 secondes de rinçage à l’eau et 5 secondes de séchage avant chaque prélèvement).
Les lames imprimées peuvent être conservées plusieurs mois dans une boîte en plastique propre (à fond non tapissé de mousse ou de liège car il a été montré que cela attaque parfois le revêtement chimique des lames) dans un endroit frais et sec (mais pas dans un réfrigérateur pour éviter la formation de condensation).
2.3. Coordonnées des gènes sur la matrice imprimée
Il est nécessaire à cette étape de pouvoir établir avec certitude l’emplacement exact de chaque gène dans la matrice des spots déposés sur la lame. Le logiciel de pilotage du robot permet en principe de convertir une liste de gènes comportant leurs coordonnées sur la plaque 384 puits en une liste avec leurs coordonnées sur la lame, mais la conversion n’est pas toujours intuitive et il faut considérer attentivement la manière dont la matrice de spots à été conçue (Figure 6). Des logiciels tels que Arraymaker8 permettent également de convertir des listes de gènes ordonnées en plaques 96 puits en une liste ordonnée en plaques 384 puits.
A1 A2 A3
B1 B2 B3
A1 A2B1 B2 A1 A2B1 B2A3 B3 A3 B3A4 B4 A4 B4A4
B4
…
…
… …
…
65 Plaques de PCR 96 puits 17 Plaques de spotting 384 puits Matrice d’impression à 4 x 4 pointes
…
C1 C2 C3
D1 D2 D3
C4
D4
C1 C2 C3D1 D2 D3 C4 D4
C1 C2C3 D1 D2D3 C4 D4
12 colonnes 24 colonnes
15 colonnes
8 lignes
16 lignes
15 lignes
Figure 6 : Etapes de transfert permettant de suivre les coordonnées de chaque gène dans les plaques PCR et dans la matrice d’impression des puces.
8 ArrayMaker Version 2 disponible sur le site http://derisilab.ucsf.edu/arraymaker.shtml.

23
3. Traitement de finition et préhybridation des lames
Le traitement des lames après le dépôt est une étape cruciale, qui permet notamment de bloquer les charges laissées libres sur la surface autour des spots. Une mauvaise réalisation conduit à une forte détérioration de la qualité des images, avec une augmentation du bruit de fond et la formation de « comètes » et/ou de « doughnuts » au niveau des spots (cf. Guide de diagnostic des anomalies, p.46). Le traitement requis varie selon la nature des lames et certaines étapes sont supposées optionnelles ou réalisées différemment selon les habitudes des laboratoires. Les étapes communes de ce traitement sont :
la réhydratation (optionnelle) des spots par de la vapeur d’eau chaude ou de tampon, suivie d’un séchage rapide sur une plaque chauffante, afin de permettre l’homogénéisation du dépôt d’ADN à l’intérieur de chaque spot ;
la fixation de l’ADN à la surface de la lame (requise pour les puces à oligonucléotides) par irradiation aux rayons UV (« cross-linking », formation de liaisons covalentes entre les résidus thymidyl et les atomes de carbone des groupements aminés) ;
la réduction des charges positives restées libres dans une solution d’anhydride succinique (optionnelle pour les lames d’amino-silane, mais recommandée pour réduire le bruit de fond) ;
la dénaturation de l’ADN dans de l’eau bouillante ou de l’éthanol à –20°C (optionnelle) ;
la préhybridation dans une solution de BSA pour diminuer l’hybridation non spécifique (requise pour les lames d’amino-silane, optionnelle pour les lames de polylysine).
Le traitement doit être effectué de préférence le jour de l’hybridation. Les lames traitées ne se conservent que 2-3 jours. Le protocole détaillé fourni en annexe (Protocole 2, p.51) est celui que nous utilisons pour traiter les lames Corning GAPS II. Il a été adapté à partir des protocoles du laboratoire DeRisi9 et du manuel du fournisseur Corning.
9 Protocole du site http://www.microarrays.org

24
III. Préparation des cibles et hybridation des puces
1. Préparation des échantillons d’ARN
1.1. Extraction des ARN totaux et messagers
La qualité de l’extraction des ARN est primordiale pour la réussite de l’expérience. Une mauvaise purification peut être la cause d’une synthèse inefficace des cibles marquées et d’une augmentation des bruits de fond sur la lame. L’ARN est aussi très sensible à la dégradation, celle-ci pouvant induire l’obtention de données biaisées en altérant la proportion des espèces présentes initialement dans l’échantillon étudié. Il est nécessaire de travailler très rapidement en portant des gants et de maintenir les tubes au froid afin d’éviter la dégradation des ARN. La surface de travail et le matériel doivent être parfaitement nettoyés afin d’empêcher la contamination par des RNases. Toutes les solutions aqueuses sont préparées avec de l’eau stérile préalablement traitée par le DEPC (1/1000e) qui inhibe les RNases. Enfin nous utilisons, à la place de l’éthanol absolu, de l’éthanol à 95% réservé à la spectrométrie UV, qui ne contient pas de résidu fluorescent. Il est possible de synthétiser les cibles fluorescentes soit directement à partir d’un extrait d’ARN totaux, soit à partir d’un extrait d’ARNm (ARN polyadénylés). La purification des ARNm nécessite d’effectuer une étape supplémentaire, qui est délicate et peut conduire à des pertes de rendements, mais qui présente plusieurs avantages :
L’extraction des ARNm offre un degré de purification supplémentaire. Les contaminants (protéines, polysaccharides, sels, phénol, ADN), qui affectent les étapes ultérieures et favorisent la dégradation de l’ARN, sont plus facilement éliminés.
L’enrichissement en ARNm nous permet d’utiliser des amorces de séquences aléatoires (« random priming »), en plus d’une amorce oligo(dT) pour synthétiser la cible cDNA marquée. L’efficacité de la transcription inverse est ainsi nettement améliorée et des cibles plus représentatives de l’ensemble des ARNm sont générées, comparées aux cibles produites seulement à partir d’amorces oligo(dT), qui sont le plus souvent tronquées et réduites à la région 3’ des transcrits.
Nous avons envisagé la possibilité d’utiliser des amorces de séquences aléatoires même avec des ARN totaux, en supposant que les cibles issues d’ARN non messagers ne s’hybrident pas sur la puce si elle ne contient pas de séquences qui leur sont complémentaires. Cependant, outre le fait que le risque d’hybridation non-spécifique augmente, il est probable que les ARN totaux comportent une proportion importante d’ARN immatures ou dégradés, qui ne sont pas représentatifs de l’expression génique spécifique à la condition étudiée. Nous avons effectivement constaté que l’hybridation d’une cible synthétisée à partir des ARN totaux, comparée à celle synthétisée à partir des

25
ARNm d’un même échantillon cellulaire, montre de grandes différences de signaux sur les spots (plus de 400 spots avec un ratio des signaux d’hybridation supérieur à 2. Pour les méthodes de mesure de l’expression différentielle, cf. §IV.3.1, p.36).
Compte tenu des remarques ci-dessus, nous préférons procéder systématiquement à une extraction des ARNm. A cet effet, nous isolons dans un premier temps les ARN totaux selon un protocole que nous avons adapté d’une méthode classique d’extraction au phénolchloroforme acide à chaud (Protocole 3, p.52). Chez la levure, et particulièrement pour les cellules en sporulation, il est important de noter que les parois cellulaires sont difficiles à lyser et doivent être broyées par une forte agitation au vortex en présence de billes de verre (contrairement à ce qui est généralement recommandé pour extraire les ARN des cellules d’autres organismes). Nous utilisons ensuite 500-1000 µg d’ARN totaux (nous avons constaté que le rendement baisse fortement avec moins de 500 µg) pour réaliser l’extraction d’ARNm. grâce au kit Micro-FastTrack (Invitrogen), qui permet de réaliser une chromatographie d’affinité sur billes d’oligo(dT)-cellulose. (Protocole 4, p.53, adapté à partir du manuel du fournisseur).
1.2. Mesure de la concentration et contrôle de la qualité des ARN
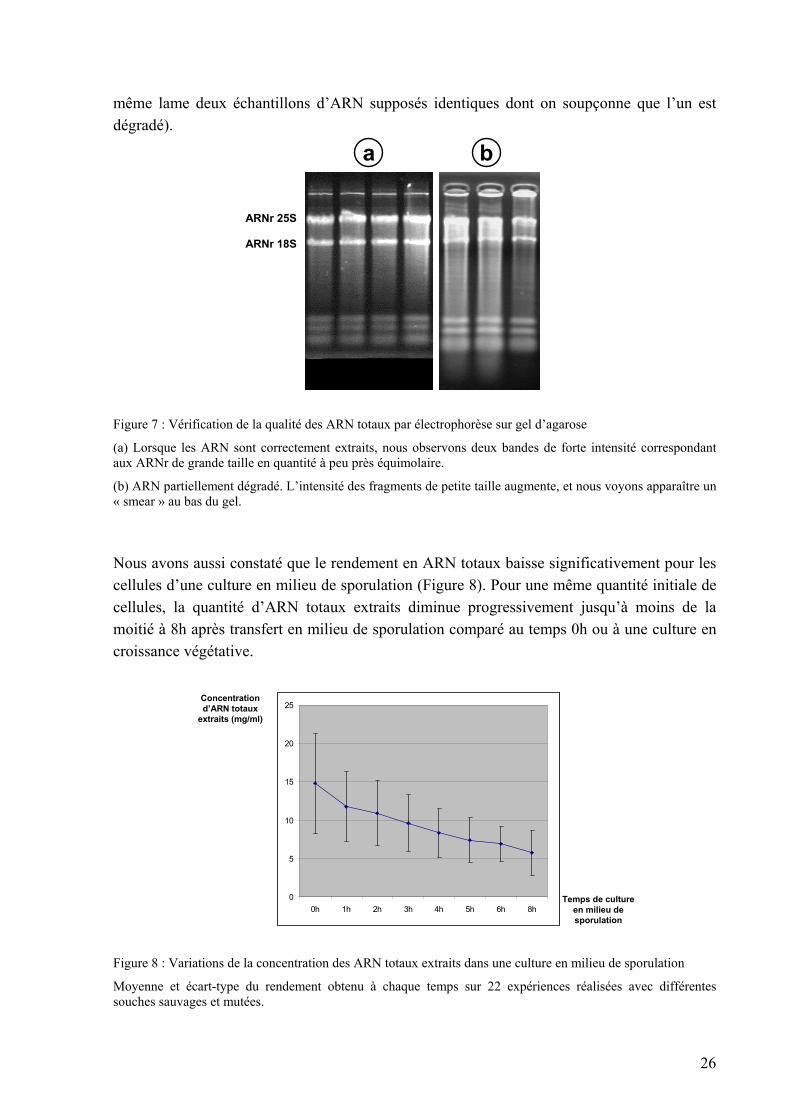
La concentration des ARN totaux et messagers après extraction est estimée en mesurant l’absorbance optique à 260 nm au spectrophotomètre : 1 A260 = 40 µg/µl d’ARN. Les rendements attendus avec les protocoles précédents sont de 500-1000 µg d’ARN totaux à partir de 1.108 cellules de levure en croissance végétative ou 5.108 cellules d’une culture méiotique, et de 10-20 µg d’ARN messagers. On estime que le rapport A260/A280 doit être supérieur à 1,8 si l’extrait d’ARN est correctement purifié. Nous considérons ce paramètre essentiellement pour détecter les problèmes d’extraction des ARNm. Les ARN totaux montrent généralement des rapports inférieurs car ils sont moins bien purifiés avec le protocole que nous utilisons, mais nous n’en tenons pas compte sachant que nous poursuivons toujours la purification avec l’extraction des ARNm. Nous vérifions également le profil des ARN totaux visualisé après électrophorèse sur un gel d’agarose et coloration au bromure d’éthydium (Figure 7). Chez la levure, nous observons deux bandes d’intensité égale à 3,3 kb (ARNr 25S) et 1,6 kb (ARNr 18S), ainsi que plusieurs bandes de faible taille autour de 0,1 kb (ARNr 5S, ARNt…). Les ARNm ne représentent qu’environ 1% de l’ensemble et forment une trace étalée (« smear ») autour de 1 kb. Une dégradation partielle se traduit par l’apparition de fragments de faible taille moléculaire. Dans ce cas, nous recommençons l’extraction car une telle dégradation visible à l’œil conduit à l’obtention de données biaisées (la dégradation n’est pas linéaire et nous avons observé que les ARN de grandes tailles tendent à être plus rapidement dégradés en hybridant sur une
26
même lame deux échantillons d’ARN supposés identiques dont on soupçonne que l’un est dégradé).
ARNr 25S
ARNr 18S
ba
Figure 7 : Vérification de la qualité des ARN totaux par électrophorèse sur gel d’agarose
(a) Lorsque les ARN sont correctement extraits, nous observons deux bandes de forte intensité correspondant aux ARNr de grande taille en quantité à peu près équimolaire.
(b) ARN partiellement dégradé. L’intensité des fragments de petite taille augmente, et nous voyons apparaître un « smear » au bas du gel.
Nous avons aussi constaté que le rendement en ARN totaux baisse significativement pour les cellules d’une culture en milieu de sporulation (Figure 8). Pour une même quantité initiale de cellules, la quantité d’ARN totaux extraits diminue progressivement jusqu’à moins de la moitié à 8h après transfert en milieu de sporulation comparé au temps 0h ou à une culture en croissance végétative.
0
5
10
15
20
25
0h 1h 2h 3h 4h 5h 6h 8hTemps de culture
en milieu de sporulation
Concentration d’ARN totaux
extraits (mg/ml)
Figure 8 : Variations de la concentration des ARN totaux extraits dans une culture en milieu de sporulation
Moyenne et écart-type du rendement obtenu à chaque temps sur 22 expériences réalisées avec différentes souches sauvages et mutées.

27
Deux hypothèses expliqueraient ce phénomène couramment observé lors d’expériences réalisées sur des cultures en sporulation :
L’hypothèse classiquement admise dans les laboratoires est que les ARN sont plus difficiles à extraire à partir de cellules en sporulation, en particulier à cause d’une augmentation des taux de polysaccharides et de la formation de parois difficiles à lyser suite à la différenciation cellulaire. Cependant, cette hypothèse n’explique pas que ce phénomène de baisse de rendement en ARN soit aussi observé avec des mutants comme ime1∆, qui n’entrent pas en sporulation.
La deuxième hypothèse est que la quantité d’ARN totaux, et en particulier des ARN ribosomaux (ARNr), diminue dans les cellules au cours de la différenciation méiotique. Nous avons constaté que l’expression des gènes impliqués dans la synthèse protéique, notamment des gènes codant pour les sous-unités des complexes ribosomaux, est fortement réprimée après transfert des cellules en milieu de sporulation. Ceci est observé même dans les cellules ime1∆. Nous pouvons donc envisager que la transcription des ARNr est également réduite en parallèle afin de diminuer l’activité de synthèse protéique. Les ARNr constituant 80% de l’ensemble des ARN cytoplasmiques, ceci expliquerait la diminution globale du rendement en ARN totaux. Van de Peppel et al. ont également observé que la synthèse globale d’ARN diminue quand la cellule entre en phase stationnaire dans un milieu appauvri en nutriments [76].
2. Transcription inverse et incorporation des marqueurs fluorescents
Les cibles sont synthétisées par une transcription inverse des ARNm en présence d’amorces oligo(dT)15 et d’amorces aléatoires (hexamers Pd(N)6) afin d’obtenir des brins d’ADNc marqués par une molécule fluorescente. Les premières expériences avaient été réalisées en effectuant une incorporation « directe » des colorants fluorescents : la transcription inverse est réalisée en ajoutant des nucléotides couplées à un fluorochrome, les plus couramment utilisés étant les carbocyanines Cy3 et Cy5. Nous avons suivi cette méthode dans un premier temps, mais nous avons été rapidement confrontés à deux inconvénients majeurs :
- Les nucléotides couplées Cy3-dUTP et Cy5-dUTP sont extrêmement onéreux.
- Ces nucléotides ne sont pas les substrats naturels des transcriptases reverses, et leur encombrement stérique est tel que l’efficacité de l’incorporation est très faible et inégale entre la Cy3 et la Cy5.
Ainsi, une méthode d’incorporation « indirecte » lui est préférée aujourd’hui pour les analyses sur puces à ADN « spottées ». Dans cette méthode, des nucléotides portant un groupement amine réactif, l’amino-allyl dUTP (aa-dUTP), sont incorporés lors de la transcription inverse. Ceux-ci sont couplés dans un deuxième temps à des N-hydroxysuccinimidyl esters (NHS-esters) de Cy3 ou de Cy5, selon la réaction décrite par Randolph et Waggoner [77].
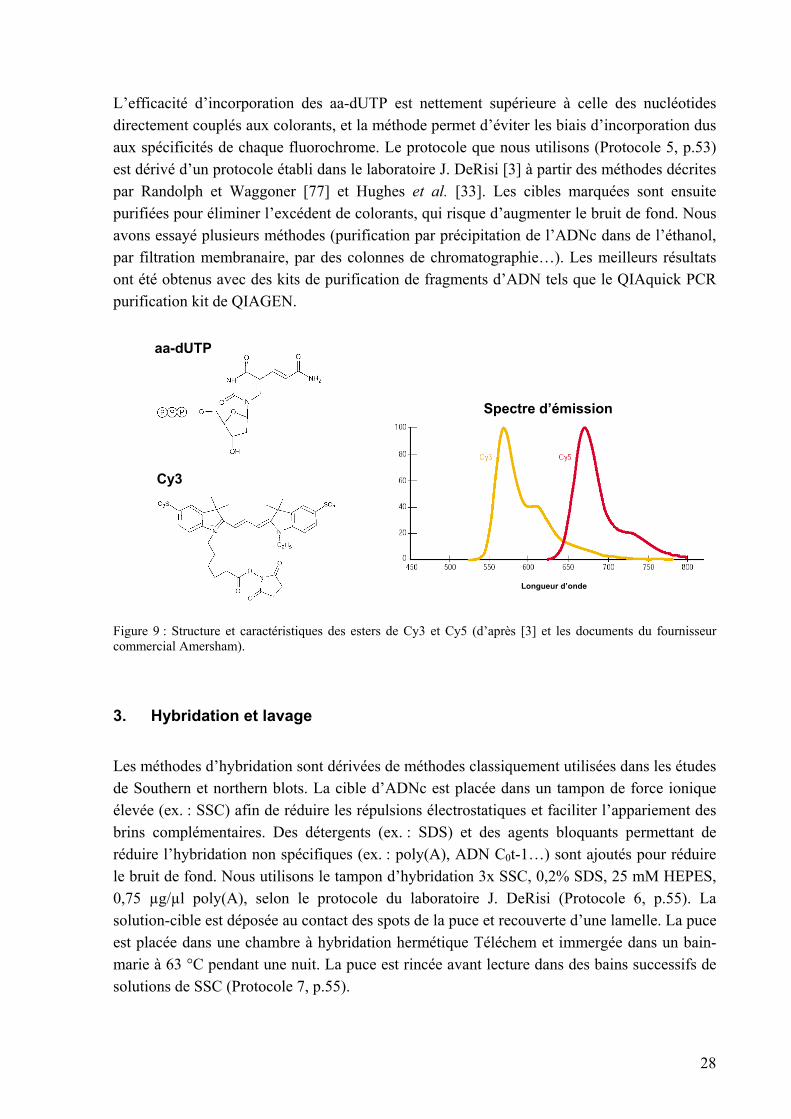
28
L’efficacité d’incorporation des aa-dUTP est nettement supérieure à celle des nucléotides directement couplés aux colorants, et la méthode permet d’éviter les biais d’incorporation dus aux spécificités de chaque fluorochrome. Le protocole que nous utilisons (Protocole 5, p.53) est dérivé d’un protocole établi dans le laboratoire J. DeRisi [3] à partir des méthodes décrites par Randolph et Waggoner [77] et Hughes et al. [33]. Les cibles marquées sont ensuite purifiées pour éliminer l’excédent de colorants, qui risque d’augmenter le bruit de fond. Nous avons essayé plusieurs méthodes (purification par précipitation de l’ADNc dans de l’éthanol, par filtration membranaire, par des colonnes de chromatographie…). Les meilleurs résultats ont été obtenus avec des kits de purification de fragments d’ADN tels que le QIAquick PCR purification kit de QIAGEN.
aa-dUTP
Cy3
Spectre d’émission
Longueur d’onde
Figure 9 : Structure et caractéristiques des esters de Cy3 et Cy5 (d’après [3] et les documents du fournisseur commercial Amersham).
3. Hybridation et lavage
Les méthodes d’hybridation sont dérivées de méthodes classiquement utilisées dans les études de Southern et northern blots. La cible d’ADNc est placée dans un tampon de force ionique élevée (ex. : SSC) afin de réduire les répulsions électrostatiques et faciliter l’appariement des brins complémentaires. Des détergents (ex. : SDS) et des agents bloquants permettant de réduire l’hybridation non spécifiques (ex. : poly(A), ADN C0t-1…) sont ajoutés pour réduire le bruit de fond. Nous utilisons le tampon d’hybridation 3x SSC, 0,2% SDS, 25 mM HEPES, 0,75 µg/µl poly(A), selon le protocole du laboratoire J. DeRisi (Protocole 6, p.55). La solution-cible est déposée au contact des spots de la puce et recouverte d’une lamelle. La puce est placée dans une chambre à hybridation hermétique Téléchem et immergée dans un bain-marie à 63 °C pendant une nuit. La puce est rincée avant lecture dans des bains successifs de solutions de SSC (Protocole 7, p.55).

29
IV. Acquisition et analyse des données
1. Acquisition des données brutes
1.1. Lecture sur le scanner
L’acquisition des images est réalisée par lecture des puces sur des scanners GenePix 4000 (Axon Instruments) installés à l’ENS (Figure 10). Le scanner est muni de deux lasers (excitations à 532 nm et 635 nm) qui permettent l'acquisition simultanée des signaux émis par les fluorochromes Cy3 et Cy5. Il permet de lire une lame complète du génome de la levure en moins de 5 mn avec une résolution de 10 µm/pixel et une sensibilité maximale de 0,1 molécule de fluor/µm2.
Lecture de la puce
Cy5>Cy3 Cy5=Cy3 Cy5<Cy3
Image en Cy5 Image en Cy3
Extraction des données
Surface prise en compte pour le calcul des
intensités des signaux rouge et vert sur le spot
Surface prise en compte pour le calcul du bruit de fond local
Figure 10 : Acquisition des données sur le scanner Genepix Pro.
Les images obtenues pour chaque canal Cy3 et Cy5 sont enregistrées au format TIFF 16 bits en 65535 niveaux de gris (enregistrement de 216-1 niveaux maximum par pixel). La gamme d’intensité détectée est donc comprise entre 0 (noir) et 65535 (blanc). La puissance de l’excitation laser doit être réglée manuellement (sur une échelle de 0 à 1000) de manière à ce que le signal sur les spots soit détectable au-dessus du bruit de fond mais ne sature pas (dans la pratique, nous réglons la puissance de manière à voir apparaître juste 2-3 spots saturés en blanc). Il est admis qu’à l’intérieur de cette gamme, l’intensité du signal augmente de manière linéaire avec la quantité de molécules de fluor sur le spot. Il faut noter que si le marquage est trop faible et requiert un niveau excessivement élevé de la puissance du laser (au-delà de 800-

30
850 selon le constructeur), l’intensité du bruit de fond augmente plus vite que celle du signal sur les spots. Le rapport signal/bruit de fond devrait être supérieur à 1,5-2 pour que le signal soit quantifiable. J’ai également utilisé, au début de ma thèse, les scanners ScanArray 3000 (le premier scanner de notre plate-forme, installé en 1999 à l’ESPCI) et ScanArray 5000 (installé à l’Institut Curie) de GSI Lumonix (aujourd’hui Perkin-Elmer). Cependant nous utilisons aujourd’hui exclusivement les scanners GenePix car ils présentent deux avantages indéniables pour une qualité d’image équivalente :
- la rapidité de lecture (les scanners ScanArray requièrent 4 fois plus de temps de lecture, soit près de 20 mn pour une lame du génome de la levure).
- la convivialité (le réglage des paramètres de lecture est plus simple avec les scanners GenePix et le logiciel intégré d’acquisition/d’extraction des données présenté ci-dessous est très performant).
1.2. Extraction des données numériques
Les images sont analysées grâce au logiciel GenePix Pro 4 (Axon Instruments) afin d’extraire les données numériques correspondant à chaque spot (Figure 10). Les images sont colorées artificiellement – celle du canal Cy3 en vert et celle de Cy5 en rouge – et superposées pour leur visualisation. Ainsi, un spot de couleur verte indique un gène dont le niveau d'expression est plus élevé dans l'échantillon marqué avec le Cy3 que celui marqué avec le Cy5, et inversement pour un spot de couleur rouge. Le spot apparaît jaune lorsque le gène est exprimé de manière identique dans les deux échantillons comparés. Le logiciel permet de définir une grille sur l’image afin d’identifier chaque spot en lui assignant des coordonnées uniques (adressage des spots, ou « gridding ») et de délimiter la surface du spot par rapport au reste de la lame (« segmentation » du « foreground » contenant le spot par rapport au « background » définissant le bruit de fond). GenePix Pro intègre un algorithme performant de placement automatique des grilles, ce qui accélère considérablement l’analyse par rapport à d’autres logiciels tels que ScanAlyse (Stanford University), pour lequel la position et la taille des cercles délimitant les spots doivent être ajustées individuellement. Le logiciel génère les données numériques correspondant aux valeurs moyennes et médianes du signal émis par les pixels de chaque spot et du bruit de fond local (délimités par la grille comme illustré dans la Figure 10) et divers autres paramètres (l’écart-type associé aux intensités des pixels d’un spot, le rapport signal/bruit de fond, etc.).

31
Rr/v(i) =
2. Transformation et stockage des données
2.1. Filtrage primaire des données brutes
Le logiciel d’extraction permet de repérer visuellement les spots non exploitables (zones de la lame couvertes de bruit de fond, spot absent…) par un système de balisage (dépôt de drapeaux ou « flags » en anglais) qui assigne un code numérique selon la qualité du spot considéré, afin de faciliter le filtrage des données non significatives (dans GenePix Pro, 0 correspond par défaut à un spot exploitable, –100 à un spot défini « mauvais », –50 à un spot non détecté par le logiciel et –25 à un spot défini « vide » par l’utilisateur). Dans Gene Pix Pro, une visualisation des paramètres calculés sur des graphes en nuages de points permet, par le même système de balisage, de filtrer les spots selon des critères personnalisés. Notamment, nous choisissons en général d’éliminer de la suite de l’analyse les spots dont la somme des intensités médianes rouge et verte est inférieure à 300, et les spots dont le rapport signal/bruit de fond est inférieur à 1,5.
2.2. Le calcul des ratios d’expression
Le niveau d’expression relatif de chaque gène est estimé par la valeur du ratio d’expression calculé à partir des intensités des signaux en rouge et en vert : intensité médiane du spot i en rouge – médiane du bruit de fond local en rouge
intensité médiane du spot i en vert – médiane du bruit de fond local en vert Ainsi le ratio est >1 si le gène i est plus exprimé dans l’échantillon marqué en rouge que celui marqué en vert et vice-versa. Il existe plusieurs méthodes de calcul du ratio d’expression (ratio des intensités moyennes, médianes, régression linéaire…). Nous avons choisi d’utiliser le ratio des intensités médianes car celles-ci sont moins sensibles que les intensités moyennes aux biais provoqués par les valeurs extrêmes (par exemple, un pixel émettant un signal particulièrement intense à cause d’une poussière déposée dessus). Pour manipuler et analyser les données, les ratios d’expression sont usuellement transformés dans une échelle logarithmique : log2(Rr/v). Le logarithme permet de transformer le ratio d’expression en une fonction linéaire, symétrique de – l’infini à + l’infini, centrée sur 0 et additive, ce qui facilite les analyses statistiques (notamment, on ne peut calculer les moyennes arithmétiques des ratios de plusieurs expériences que dans un espace logarithmique). On a ainsi log(Rr/v) = -log(1/ Rr/v). N’importe quelle base de logarithme peut être utilisée de manière équivalente. Nous utilisons le logarithme de base 2, qui est commode pour se rendre compte facilement de la vraie valeur du ratio (log2(2) = 1 ; log2(4) = 2 ; log2(8) = 3 ; etc.).

32
2.3. La normalisation
La normalisation consiste à ajuster l’intensité globale des images acquises sur chacun des deux canaux rouge et vert, de manière à corriger des biais techniques systématiques qui tendent à déséquilibrer le signal de l’un des canaux par rapport à l’autre. Ces biais sont dus en particulier aux différences de caractéristiques des deux fluorochromes Cy3 et Cy5, qui ne possèdent pas le même coefficient d’extinction molaire (à incorporation égale, Cy5 émet un signal plus fort que Cy3), aux différences d’incorporation des marqueurs lors de la synthèse des cibles, et aux paramètres de lecture au scanner (réglages de la puissance des lasers…). Il existe plusieurs méthodes pour normaliser les données.
2.3.1 La normalisation par rapport à la moyenne globale des intensités
On suppose que la majorité des gènes sont exprimés de la même manière dans les deux échantillons comparés, et donc que la majorité des spots émettent un signal d’intensité égale en rouge et en vert. Dans ces conditions, la moyenne arithmétique des ratios d’expression Rr/v de tous les spots devrait être égale à 1. Si tel n’est pas le cas, nous corrigeons les ratios d’expressions par un facteur de normalisation N, tel que : Pour chaque gène : Rr/v normalisé = N x Rr/v
où N = 1/exp(moyenne de log2(Rr/v) de tous les spots de la puce) Afin d’éviter les biais, les spots non exploitables (éliminés par les filtres) et les spots ayant des intensités rouges et vertes très différentes (Rr/v >10 ou < 0,1 dans GenePix Pro) ne sont pas pris en compte dans le calcul du facteur de normalisation. On peut aussi utiliser la médiane des ratios à la place de la moyenne.
2.3.2 La normalisation par rapport à des spots témoins
Cette deuxième méthode consiste à calculer le facteur de normalisation N, non pas à partir de l’ensemble des spots de la lame, mais d’un nombre restreint de spots définis, dont on sait qu’ils émettent un même signal en rouge et en vert. Il peut s’agir de témoins de normalisation préétablis (ex : ADN génomique à différentes concentrations) ou de « house-keeping genes » qui sont supposés être exprimés de manière constante dans les conditions de l’expérience. Cette méthode est sans doute préférable à la précédente dans les conditions où il y a beaucoup de gènes exprimés de manière différentielle et si la distribution des ratios d’expression est asymétrique. Cependant, il n’est pas toujours aisé de définir des témoins de normalisation adaptés aux conditions de l’étude et d’être certain que l’expression de ces témoins ne varie pas au cours de l’expérience.
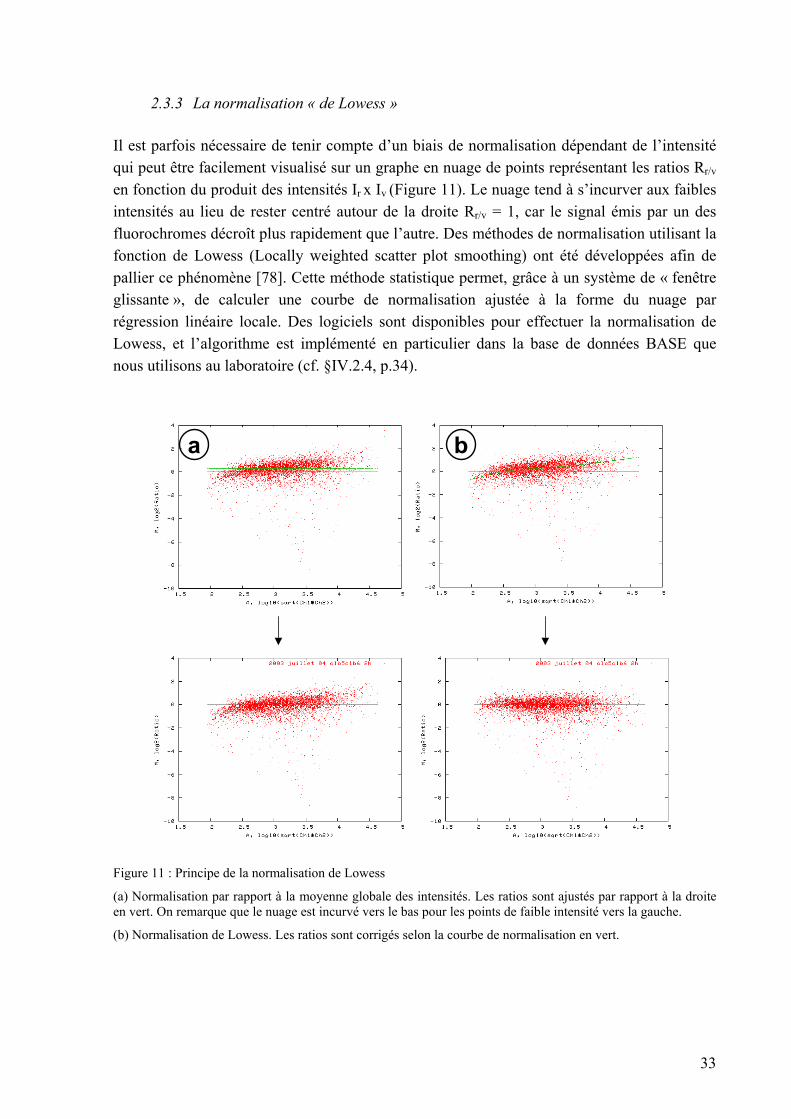
33
2.3.3 La normalisation « de Lowess »
Il est parfois nécessaire de tenir compte d’un biais de normalisation dépendant de l’intensité qui peut être facilement visualisé sur un graphe en nuage de points représentant les ratios Rr/v en fonction du produit des intensités Ir x Iv (Figure 11). Le nuage tend à s’incurver aux faibles intensités au lieu de rester centré autour de la droite Rr/v = 1, car le signal émis par un des fluorochromes décroît plus rapidement que l’autre. Des méthodes de normalisation utilisant la fonction de Lowess (Locally weighted scatter plot smoothing) ont été développées afin de pallier ce phénomène [78]. Cette méthode statistique permet, grâce à un système de « fenêtre glissante », de calculer une courbe de normalisation ajustée à la forme du nuage par régression linéaire locale. Des logiciels sont disponibles pour effectuer la normalisation de Lowess, et l’algorithme est implémenté en particulier dans la base de données BASE que nous utilisons au laboratoire (cf. §IV.2.4, p.34).
a b
Figure 11 : Principe de la normalisation de Lowess
(a) Normalisation par rapport à la moyenne globale des intensités. Les ratios sont ajustés par rapport à la droite en vert. On remarque que le nuage est incurvé vers le bas pour les points de faible intensité vers la gauche.
(b) Normalisation de Lowess. Les ratios sont corrigés selon la courbe de normalisation en vert.

34
2.4. Le stockage dans des bases de données et le suivi des expériences
La multiplication des études de transcriptomes induit l’accumulation rapide d’une masse considérable de données à traiter. Par exemple, une expérience sur une seule lame de puce de levure génère différentes valeurs numériques sur chacun des 6200 ORFs du génome (intensités moyennes et médianes, intensité du bruit de fond, estimation de l’erreur associée aux mesures…). Une dizaine de lames sont généralement requises pour une étude cinétique complète d’un processus biologique. On imagine ainsi aisément qu’un projet de comparaison de différentes cinétiques nécessite plusieurs dizaines voire centaines de lames et génère en conséquence un accroissement d’ordre exponentiel des données. La gestion des données est ainsi devenue un enjeu crucial pour tout projet d’analyse de transcriptome. Plusieurs types de données doivent être stockés de manière accessible tout au long du projet :
Les images TIFF originelles, acquises par le scanner. Il est indispensable de conserver ces fichiers pour pouvoir revenir aux données initiales, mais l’inconvénient est que leur taille est très importante (environ 32 Mo pour une lame complète de levure scannée à une résolution de 10 µm). A ce jour, nous enregistrons une copie des images sur un disque dur externe puis nous les gravons sur un DVD-Rom pour une sauvegarde à long terme.
Les tableaux de données brutes et transformées. Les données numériques avant et après transformations (filtrage, normalisation, traitements statistiques…) doivent être disponibles et aisément manipulables.
Les informations relatives aux expériences. Il est important de noter que les résultats dépendent des conditions expérimentales (conditions de culture, de synthèse de cibles…) et de fabrication des lames (qualité des produits de PCR déposés, du spotting…). Toutes ces informations doivent être consignées dans un système de gestion adapté (« LIMS » = «Laboratory Information Management System ») pour permettre une traçabilité complète.
Les informations biologiques relatives aux gènes étudiés. Pour faciliter l’interprétation, outre une identification claire de chaque spot et un accès aux bases de données externes sur le réseau Internet, il est souhaitable de disposer d’un minimum d’annotations directement associées aux données au cours de l’analyse.
Nous avons successivement utilisé les bases de données AMAD (Another Microarray Database) et BASE (BioArray Software Environment) pour gérer les résultats de notre laboratoire. Ces deux logiciels, distribués sur Internet en code source libre, fonctionnent sur un ordinateur sous système Unix (Linux) qui fait office de serveur, de manière à ce que la base puisse être consultée et manipulée à partir d’un navigateur Internet de n’importe quel autre ordinateur connecté sur le réseau.

35
2.4.1 AMAD
AMAD10, développée par l’équipe M. Eisen, J. DeRisi, P. Spellman et M. Diehen à Stanford University, est une base relativement simple à installer et à utiliser, qui permet de soumettre des fichiers de résultats générés par les logiciels Scanalyse ou Genepix, d’effectuer des opérations de filtrage, de normalisation par la médiane globale des intensités, et de récupérer les données individuellement ou groupées par séries d’expériences. Les inconvénients sont que son système de classement est mal adapté à la gestion d’un grand nombre d’expériences, qu’il manque de souplesse comme il ne s’agit pas d’une base de données relationnelle (en particulier, il est difficile de corriger une information, telle que le nom d’un gène de la puce, indépendamment du reste, ce qui nous oblige à re-soumettre tous les fichiers si nous souhaitons apporter la moindre modification) et qu’il n’est plus en développement depuis quelques années. Une nouvelle version appelée NOMAD 11 est développée au laboratoire DeRisi, mais nous ne l’avons pas essayée car elle n’était pas encore achevée quand nous avons commencé à utiliser BASE.
2.4.2 BASE
BASE12 est une base relationnelle très complète de l’équipe de C. Peterson à Lund University qui bénéficie d’un développement particulièrement actif [79]. Des mises à jour avec de nouvelles fonctionnalités sont publiées plusieurs fois par an (la version la plus récente que j’ai installée au laboratoire au moment de la rédaction de ce mémoire est la 1.2.9). De plus, le logiciel dispose d’un système de modules (« plug-in ») que les développeurs peuvent ajouter indépendamment pour l’adapter à différents besoins. Ainsi, des contributeurs externes peuvent proposer d’intégrer de nouveaux systèmes de traitement de données qui n’existent pas dans la base originelle. BASE permet de stocker les fichiers de résultats issus de différents types de plates-formes et de les lier à divers paramètres expérimentaux, de protocoles et de la fabrication des lames. Elle dispose d’un système de requête et de filtrage efficace, de plusieurs options de visualisation des données (graphes en nuages de points, étiquettes de couleurs…) et de modules permettant d’effectuer différents types de normalisation et d’analyses statistiques. Les données peuvent être extraites sous divers formats compatibles avec les logiciels d’analyses les plus usités. Les inconvénients constatés sont que nous ne pouvons pas entrer d’annotation supplémentaires au niveau des gènes et que nous pouvons soumettre des images au format compressé JPEG mais pas les images TIFF d’origine. Un point à mentionner est que 10 AMAD disponible sur le site http://www.microarrays.org/software.html 11 Site de NOMAD : http://ucsf-nomad.sourceforge.net/ 12 Site de BASE : http://base.thep.lu.se/

36
l’installation et la mise à jour de BASE n’est pas conviviale et nécessite des connaissances sur l’environnement Linux et les systèmes de gestion de bases relationnelles tels que MySQL. Il serait nécessaire pour un laboratoire qui souhaite s’impliquer dans des projets d’analyse de transcriptomes d’une certaine envergure et à long terme de disposer d’un administrateur du serveur de données qui puisse maintenir la base, effectuer les sauvegardes de secours, etc.
3. Analyse et interprétation des résultats
3.1. La mesure de l’expression différentielle
Les puces à ADN permettent de mesurer le niveau d’expression relatif de chaque gène dans un échantillon cellulaire comparé à un contrôle de référence. Plus le ratio d’expression Rr/v est élevé, plus la quantité d’ARNm du gène est importante dans l’échantillon considéré. Classiquement, on considère que le niveau d’expression du gène est significativement plus élevé dans l’échantillon par rapport à la référence si Rr/v ≥ 2 (soit log2(Rr/v)=1), et inversement qu’il est significativement réduit si Rr/v ≤ 0,5 (soit log2(Rr/v)=-1). Ce seuil a été originellement établi en supposant que les ratios d’expression ont une distribution aléatoire suivant la loi normale, auquel cas l’intervalle de confiance à 95% de la valeur du ratio est de ± 1,96 (soit environ ± 2). Dans la pratique, des seuils de confiance sont fixés de manière arbitraire pour déterminer si le ratio indique une variation d’expression significative. Certains laboratoires recommandent de calculer un seuil différent à chaque expérience en fonction de la moyenne et de l’écart-type de l’ensemble des ratios dans l’expérience considérée. Cependant, cette méthode n’est applicable que si le nombre de gènes dont l’expression varie fortement est relativement faible par rapport à l’ensemble de la population permettant d’établir la variabilité aléatoire des ratios. Enfin, il est souvent recommandé de reproduire l’expérience plusieurs fois avec des échantillons indépendants afin d’estimer la variabilité technique et biologique pour distinguer l’effet réel étudié des fluctuations aléatoires. Il serait sans doute possible de considérer des seuils plus proches de 1 avec un nombre suffisant d’expériences répétées indépendamment (par exemple, un gène montrant systématiquement un ratio de 1,5 aurait plus de chances d’être transcrit préférentiellement qu’un autre dont le ratio varierait aléatoirement entre 2 et 0,5). Il est toutefois difficile de réaliser des études de reproductibilité à grande échelle en raison notamment des limitations matérielles et financières. La Figure 12 illustre le type de variations du profil d’expression que nous pouvons observer. Sur ces graphes, chaque point correspond à un gène. Nous avons reporté en ordonnée le logarithme du rapport des signaux rouge sur vert (log2(Rr/v)) et en abscisse le logarithme de la somme des intensités (log2(Ir+Iv)).
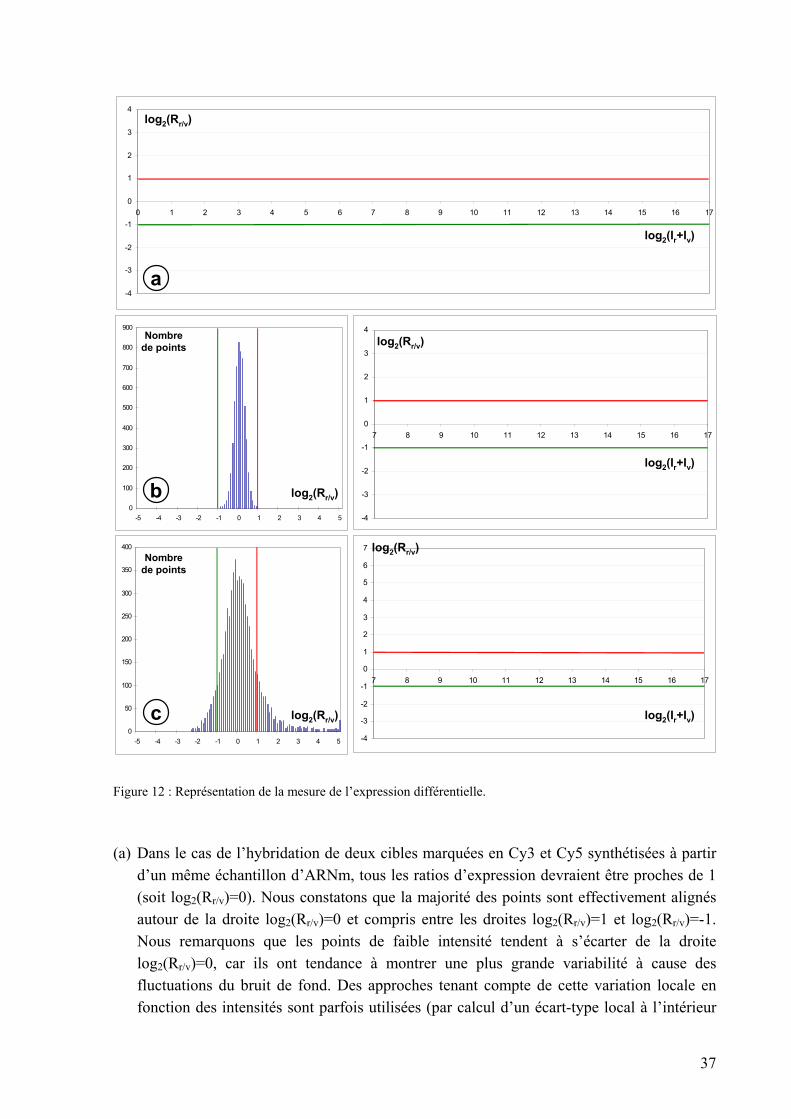
37
0
50
100
150
200
250
300
350
400
-5 -4 -3 -2 -1 0 1 2 3 4 5
0
100
200
300
400
500
600
700
800
900
-5 -4 -3 -2 -1 0 1 2 3 4 5 -4
-3
-2
-1
0
1
2
3
4
7 8 9 10 11 12 13 14 15 16 17
-4
-3
-2
-1
0
1
2
3
4
5
6
7
7 8 9 10 11 12 13 14 15 16 17
-4
-3
-2
-1
0
1
2
3
4
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17
a
b
c
log2(Rr/v)
log2(Ir+Iv)
Nombre de points
log2(Rr/v)
log2(Rr/v)
log2(Rr/v)
log2(Rr/v)
log2(Ir+Iv)
log2(Ir+Iv)
Nombre de points
Figure 12 : Représentation de la mesure de l’expression différentielle.
(a) Dans le cas de l’hybridation de deux cibles marquées en Cy3 et Cy5 synthétisées à partir
d’un même échantillon d’ARNm, tous les ratios d’expression devraient être proches de 1 (soit log2(Rr/v)=0). Nous constatons que la majorité des points sont effectivement alignés autour de la droite log2(Rr/v)=0 et compris entre les droites log2(Rr/v)=1 et log2(Rr/v)=-1. Nous remarquons que les points de faible intensité tendent à s’écarter de la droite log2(Rr/v)=0, car ils ont tendance à montrer une plus grande variabilité à cause des fluctuations du bruit de fond. Des approches tenant compte de cette variation locale en fonction des intensités sont parfois utilisées (par calcul d’un écart-type local à l’intérieur

38
d’une « fenêtre glissante » de type fonction de Lowess). Cependant, comme précédemment, cette méthode n’est valable que pour des données comportant une majorité de ratios proches de 1, et seulement un faible nombre de gènes exprimés de manière différentielle.
(b) Nous éliminons systématiquement les points correspondant aux spots présentant des
défauts d’impression et des intensités très faibles, susceptibles de montrer une variation de ratio trop importante (soit, dans cet exemple, les points ayant une somme des intensités inférieure à 300). Après ce filtrage, très peu de points sont à l’extérieur des droites log2(Rr/v)=1 et log2(Rr/v)=-1. L’histogramme à gauche représente la distribution du nombre de points en fonction du ratio et met en évidence une distribution symétrique, étroitement centrée autour de 0. Afin de simplifier, nous avons donc considéré que nous pouvons définir un seuil de confiance fixe log2(Rr/v)=±1 pour la suite des analyses.
(c) L’hybridation sur une puce de cibles préparées à partir de deux échantillons cellulaires
différents (dans cet exemple : une culture en croissance végétative comparée à une culture en milieu de sporulation) nous permet d’observer de nombreux gènes montrant des ratios supérieurs au seuil de 2 ou inférieurs à 0,5. L’altération du nuage de points et de l’élargissement de l’histogramme met en évidence l’expression différentielle d’un nombre important de gènes consécutivement à la perturbation apportée.
3.2. Le regroupement hiérarchique des profils d’expression
La comparaison des résultats issus de plusieurs expériences – par exemple, des expériences permettant de suivre les variations du transcriptome au cours du temps ou de comparer plusieurs types de cellules – requiert le développement de méthodes d’analyse facilitant l’organisation et l’exploitation des données.
3.2.1 Principe de l’analyse par regroupement des profils d’expression
Une méthode communément utilisée pour explorer les données est le regroupement (appelé « clustering » en anglais) des gènes présentant des profils d’expression similaires dans les expériences considérées. Ceci permet d’établir des groupes (« clusters ») de gènes co-régulés dans les conditions étudiées sans préjuger de leur fonction. Il existe plusieurs méthodes de regroupement (décrites et comparées notamment dans la thèse de A. Sturn, 2000 [80] : regroupement hiérarchique, Self Organizing Maps, k-means clustering, Analyse en Composantes Principales, Support Vector Machines). Ce type de regroupement est utile pour plusieurs raisons :
Les gènes impliqués dans une même fonction cellulaire sont susceptibles d’être exprimés de manière coordonnée. Notamment, plusieurs études établissent que des

39
protéines interagissant entre elles ou appartenant à un même complexe présentent des profils d’expression corrélés [81, 82]. Des hypothèses sur la fonction de gènes non caractérisée peuvent être donc émises en se référant aux fonctions connues des autres gènes co-régulés [83].
De même, les gènes dont les profils d’expression sont similaires sont susceptibles de dépendre d’un même mécanisme de régulation transcriptionnelle. Ceci peut faciliter la recherche de motifs de séquence consensus dans les promoteurs de ces gènes et des facteurs de transcription dont ils sont les cibles.
Certains groupes de gènes peuvent être caractéristiques d’une réponse à des conditions expérimentales spécifiques. Des clusters de gènes « signatures » peuvent être définis pour caractériser un effet particulier ou l’état physiologique des échantillons cellulaires (par exemple, gènes « signatures » spécifiquement induits lors de dommages à l’ADN [40] ou gènes permettant l’établissement de « cartes d’identités tumorales » [42]).
3.2.2 Le regroupement hiérarchique des profils avec Cluster et Treeview
Nous utilisons dans notre laboratoire la méthode de regroupement hiérarchique (« pair-wise hierarchical clustering ») décrite par Eisen et al. en 1998 avec le logiciel Cluster13 [83]. Les gènes sont comparés deux à deux en fonction du degré de similitude entre leurs profils d’expression. Les gènes montrant la plus faible distance entre leurs profils sont groupés sous un « nœud » (« node »). Un profil représentatif du groupe de gènes est attribué à ce nœud (profil moyen du groupe dans la méthode dite « average linkage », profil du gène le plus éloigné du groupe voisin pour la « complete linkage » ou le plus proche pour la « single linkage »). Le nœud est ensuite lui-même comparé à un autre gène ou un autre nœud. Ainsi, de proche en proche, les gènes sont ordonnés de manière hiérarchique dans un dendrogramme qui peut être visualisé avec le logiciel Treeview (exemple simplifié dans la Figure 13). Treeview permet d’afficher les profils des gènes sous forme d’une « carte d’expression » colorée (« gene expression map »). Chaque ligne représente un gène et chaque colonne une expérience. La valeur du logarithme du ratio d’expression est reportée en rouge lorsqu’elle est positive, et en vert lorsqu’elle est négative, l’intensité de la couleur étant proportionnelle à l’amplitude de la variation. La longueur des branches de l’arbre représente la distance entre chaque nœud. Les branches des arbres peuvent être permutées sans détruire la structure de la hiérarchie. Il est possible dans Cluster d’imposer l’ordre d’affichage des gènes pour faciliter la visualisation dans Treeview, par exemple en effectuant un tri préliminaire des gènes en fonction de leur ratio maximal d’expression. Il est également possible d’attribuer des « poids » (« weight ») différents aux gènes et/ou aux expériences de manière à ce que les gènes/expériences ayant le plus de poids soient d’avantage pris en compte que les autres dans le calcul des distances.
13 Cluster et Treeview disponibles sur le site http://www.microarrays.org/software.html

40
Différentes métriques sont utilisées pour calculer la distance entre les profils, comme le coefficient de Pearson, la distance euclidienne, la covariance, la distance de Manhattan, le Kendall’s tau, etc. Le coefficient de corrélation de Pearson est l’un des plus couramment employés. Il est égal à 1 si les deux profils sont identiques, à 0 s’ils sont indépendants et à –1 s’ils sont exactement opposés. Il permet d’évaluer le degré de similitude entre les profils de deux gènes en fonction de la forme de leurs courbes sans tenir compte de l’amplitude de la variation (en d’autres termes, deux gènes co-régulés induits exactement au même moment seront considérés similaires même si l’un est exprimé à un niveau plus élevé que l’autre). Nous utilisons aussi le coefficient de Pearson « non centré » qui, contrairement, tient aussi compte des différences d’amplitude.
a b
Figure 13 : Exemple de carte d’expression générée par regroupement hiérarchique des données.
(a) Données classées aléatoirement.
(b) Le regroupement en fonction du degré de similitude des profils d’expression révèle des ensembles de gènes fonctionnellement corrélés.
3.2.3 Autres logiciels d’analyse des données
Nous avons essayé deux autres logiciels plus récents permettant d’effectuer les analyses de regroupements et distribués librement sur Internet : J-Express 14 et Genesis [84] 15 . Ces logiciels sont performants, présentent une interface graphique conviviale et de nombreuses options de visualisation des résultats. Ils permettent d’effectuer facilement des analyses par différentes méthodes, en plus du regroupement hiérarchique. Cependant nous avons noté certains inconvénients avec ces logiciels :
La navigation dans les arbres générés et la visualisation des groupes de gènes est beaucoup moins aisée qu’avec Treeview.
14 Site de J-Express : http://www.molmine.com/jexpress/j_express.html 15 Site de Genesis : http://genome.tugraz.at/Software/

41
Il est impossible d’intégrer des annotations personnalisées (fonction des gènes, résultats d’autres expériences…) de manière à les visualiser en même temps que les profils d’expression.
L’interface de Genesis semble nécessiter beaucoup de mémoire informatique (fonctionnement impossible sur les ordinateurs du laboratoire avec des matrices de données comportant plus de 2000 gènes sur une dizaine d’expériences). Il faut remarquer que la méthode de regroupement hiérarchique requiert elle-même une puissance de calcul importante, le temps de calcul nécessaire augmentant proportionnellement au nombre d’expériences et au carré du nombre de gènes.
Les versions les plus récentes de J-Express sont commerciales et ne sont plus distribuées libres de droits.
Ces logiciels nous ont permis d’essayer d’autres méthodes que le regroupement hiérarchiques telles que le k-means clustering ou l’Analyse en Composantes Principales. Ceux-ci sont efficaces lorsqu’il s’agit de dégager des tendances majeures de variations, mais se sont révélés peu pratiques pour analyser finement les sous-groupes de gènes aux profils complexes qui sont générés dans le cas précis de nos études de transcriptomes méiotiques présentés dans la deuxième partie de ce document. Nous avons ainsi privilégié, pour nos analyses, l’utilisation de la méthode de regroupement hiérarchique par Cluster et Treeview. Il convient toutefois de souligner que ces méthodes d’analyse par regroupement ne sont que des outils pour organiser et visualiser les données et qu’aucune ne donne une réponse absolue pour interpréter les résultats.
3.3. La comparaison avec les données existantes
3.3.1 Les informations sur les gènes et leurs séquences
Il est important, pour interpréter les résultats, de prendre en considération les informations disponibles dans d’autres bases de données. A cet égard, le génome de la levure S. cerevisiae présente l’avantage d’être particulièrement bien annoté. Chaque gène est identifié par un nom systématique unique, indiquant sa position relative sur les chromosomes, et un nom d’usage s’il a été caractérisé (composé de trois lettres et des chiffes, souvent systématisé par rapport à la fonction ou le phénotype des mutants : par exemple SPO11 = « SPOrulation »). Des informations détaillées sont consignées dans des bases publiques telles que SGD16 [10]. SGD fait notamment partie du Gene Ontology Consortium17 [85, 86] qui vise à annoter les gènes de différents organismes modèles par des termes précisément définies et contrôlées. Le GO consortium fournit une courte description de la fonction moléculaire de chaque protéine, du procédé biologique dans lequel elle est impliquée et sa localisation cellulaire. Ces descriptions peuvent être téléchargées sous forme de tableaux et directement intégrées dans les listes des
16 Site de Saccharomyces Genome Database : http://www.yeastgenome.org/ 17 Site du GO consortium : http://www.geneontology.org/

42
résultats de nos expériences. Ainsi, dans l’exemple de la Figure 13, la carte d’expression met immédiatement en évidence des groupes de gènes co-régulés qui sont impliqués dans un même processus biologique. La mise à jour progressive des données de séquences et des annotations dans les bases de données en ligne peut induire des confusions si les données des puces à ADN ne sont pas régulièrement mises à jour en parallèle. Depuis la publication initiale de la séquence du génome de S. cerevisiae, diverses corrections y ont été apportées suite à des études de re-séquençage et à l’identification de nouveaux ORFs par des méthodes complémentaires telles que le SAGE [13], la comparaison avec les génomes d’autres espèces de levures [87-89], l’étude phénotypique de banques de mutants générés par mutagenèse aléatoire par insertion de transposons [90], ou des prédictions établies à partir de données d’analyses de l’expression transcriptionnelle et des structures de protéines [91]. Certains ORFs ont été supprimés ou fusionnés avec d’autres, ce qui rend parfois difficile la comparaison avec des données d’expériences antérieures. Ainsi, les puces que nous avons construites contenaient environ 6200 ORFs définis sur SGD à partir des données du séquençage initial du génome de la levure. Or aujourd’hui (1er trimestre 2004), suite à ces corrections, SGD définit 5794 ORFs « vérifiés » ou « non caractérisés », et environ 810 ORFs « suspects » (ORFs hypothétiques définis à partir de données non confirmées). Nous nous sommes efforcés de mettre à jour nos annotations autant que possible afin de respecter la concordance avec la base SGD.
3.3.2 La comparaison avec des analyses publiées de transcriptomes
Il est souvent intéressant de comparer les résultats à ceux issus d’autres laboratoires. Cependant il n’est pas toujours aisé de récupérer les données, même publiées, et d’établir une comparaison rigoureuse pour deux raisons :
Les données sont souvent disséminées sur différents sites Internet en tant que documents supplémentaires de publications et sont stockées dans des fichiers de formats divers et non standardisés.
Les données brutes générées dépendent des conditions expérimentales, du choix des contrôles de références, des méthodes d’analyses, etc. Il est donc difficile, voire impossible, d’établir une comparaison sans disposer de ces informations.
Certaines bases de données généralistes telles que SGD et YPD18 [12], ou spécialisées telles que GermOnline19 [92], tentent de regrouper les résultats de différentes études publiées, mais les profils d’expression ne sont fournis qu’individuellement, par gène et par expérience. La base de données yMGV20 (yeast Microarray Global Viewer) développée par P. Marc à l’ENS regroupe également les données d’environ 80 études publiées sur le transcriptome de la levure
18 Site du Proteome BioKnowledge Library de Incyte : https://www.incyte.com/control/tools/proteome 19 Site de GermOnline : http://germonline.unibas.ch/index.php 20 Site de yMGV: http://www.transcriptome.ens.fr/ymgv/

43
qui peuvent être visualisées en parallèle [93]. Néanmoins, il est souvent nécessaire de récupérer l’ensemble des données brutes pour effectuer des analyses comparatives détaillées. Afin de rationaliser la publication des résultats de puces à ADN et faciliter l’échange d’informations, un consortium a été formé en 1999 pour établir des standards d’annotation, de représentation des données et de description des expériences : le groupe de réflexion MGED21 (Microarray Gene Expression Database), réunit ainsi plusieurs institutions aussi bien académiques que privées (EBI, TIGR, Rosetta , Stanford, Harvard…). Deux projets majeurs ont vu le jour :
La définition du MIAME (Minimal Information About a Microarray Experiment) impose un ensemble minimal d’informations à fournir à la communauté scientifique, suffisant pour permettre d’interpréter et de reproduire les résultats publiés.
Le MAGEML (MicroArray Gene Expression Markup Language) a pour but la définition d’une structure permettant de communiquer les informations sur les études de puces à ADN avec un langage commun de vocabulaires contrôlés facilitant l’informatisation des données et leurs transferts.
De plus en plus d’éditeurs de journaux scientifiques exigent le respect des normes établies par le MIAME, ainsi que le dépôt des résultats expérimentaux dans des bases de données publiques. Pour nos publications, nous avons utilisé Array Express22, qui permet de mettre en ligne les données et toutes les informations complémentaires selon les formats spécifiés par le MIAME [94]. Array Express fournit, pour chaque expérience cataloguée, un numéro d’inscription (« accession number ») qui doit être indiqué sur les publications.
3.4. Réflexions sur la conception de l’expérience biologique
Comme pour toute expérience scientifique, lorsque l’on conduit un projet d’analyse de transcriptome, il est important d’établir rigoureusement les conditions de l’étude biologique que l’on souhaite réaliser afin d’obtenir des résultats significatifs. Certains aspects spécifiques à la conception des expériences sur puces à ADN nous sont apparus cruciaux et sont décrits ci-dessous.
3.4.1 Choix du paramètre étudié et de la méthode de comparaison
La conception de l’expérience dépend en premier lieu de la question biologique posée ([95] pour revue). Comme souligné précédemment, les analyses sur des puces de type « microarrays spottées » sont toujours réalisées par la comparaison d’un échantillon cellulaire à un autre pour obtenir une mesure de niveau d’expression relatif des gènes et non absolu.
21 Site de MGED : http://www.mged.org/ 22 Site de ArrayExpress : http://www.ebi.ac.uk/arrayexpress/

44
Cette contrainte inhérente à la technologie implique que les résultats dépendent du choix des deux échantillons-cibles hybridés simultanément sur la puce. La mise en œuvre expérimentale et les méthodes d’analyse ne seront pas les mêmes selon que l’on souhaite cribler des gènes impliqués dans une réponse cellulaire spécifique, suivre une variation de la réponse au cours du temps, ou comparer plusieurs mutants afin d’établir des groupes fonctionnels, etc. Je détaillerai, dans la Partie 2 de mon manuscrit, deux exemples de stratégies auxquelles nous avons eu recours pour deux applications différentes : l’analyse des variations du transcriptome au cours du temps pendant la méiose et la localisation des sites de fixation de protéines impliquées dans la formation de cassures double-brin méiotiques. En particulier, les points suivants doivent être pris en considération pour optimiser la conception initiale du projet :
Il est préférable autant que possible de n’étudier qu’une seule variable à la fois. Il est très difficile d’interpréter des données obtenues dans des conditions limites ou transitoires, ou sous l’effet de traitements ou de mutations pléiotropes. Il est ainsi souhaitable de connaître a priori, à l’aide d’autres types d’expériences de contrôle, les conditions optimales pour que la réponse étudiée se manifeste la plus clairement possible. Lorsque l’on souhaite comparer plus de deux échantillons, il est nécessaire de choisir
une référence commune permettant de normaliser les résultats de chaque hybridation. Cette référence peut être constituée de l’un des échantillons à comparer (par exemple, la souche sauvage si on compare plusieurs souches mutées, ou le témoin de contrôle pris au temps t=0h d’une étude cinétique, etc.). Cependant, comme nous le verrons dans les applications décrites dans la deuxième partie, dans certains cas particuliers il peut être plus judicieux de choisir un échantillon de référence distinct tel que de l’ADN génomique, un mélange arbitraire d’ARN, ou encore un mélange des ARN de tous les échantillons à comparer. Cette méthode offre l’avantage d’assurer l’obtention d’un signal d’hybridation significatif sur tous les spots de la puce et de permettre une stratégie plus flexible (il est notamment plus facile de constituer une réserve importante de ce mélange de référence et d’inclure par la suite des échantillons supplémentaires dans l’analyse). Enfin, il convient de tenir compte du type et de la quantité de matériel dont on
dispose (nombre de puces, facilité d’obtention des échantillons d’ARN…) et de les confronter à ce qui sera requis pour réaliser les expériences de manière optimale, y compris les contrôles et répliques nécessaires à l’obtention de données fiables. La définition de priorités expérimentales doit être considérée d’autant plus attentivement que certains choix sont imposés par les limitations matérielles et techniques.

45
3.4.2 Optimisation des conditions expérimentales
L’expression globale des gènes est très sensible aux variations des conditions expérimentales. Etant donné que l’on considère les variations transcriptionnelles de l’ensemble des gènes d’un organisme, il se peut que les effets observés rendent compte, non pas de la réponse cellulaire étudiée, mais d’artéfacts techniques ou biologiques mal maîtrisés. La reproductibilité des résultats peut en être aussi grandement affectée et il est particulièrement difficile dans ces circonstances de comparer des données d’expériences qui n’ont pas été réalisées au même moment dans les mêmes conditions, et a fortiori des données issues de deux laboratoires différents. Une attention particulière a été portée sur les points suivants : Les fonds génétiques des souches comparées doivent être identiques. Les souches de
laboratoire peuvent présenter de grandes différences aussi bien génotypiques (par rapport à la souche S288c dont le génome a été séquencé, Primig et al. ont notamment identifié 39 délétions et 2025 polymorphismes dans la souche SK1 et 8 délétions et 318 polymorphismes souche W303 [38]) que phénotypiques (par exemple, les souches SK1 sporulent beaucoup plus rapidement et plus efficacement que les autres). Les souches mutantes analysées devraient être isogéniques à la souche sauvage de contrôle et la différence ne porter que sur la mutation étudiée. Il convient d’homogénéiser les marqueurs d’auxotrophies entre les souches à comparer, ainsi que le type sexuel s’il s’agit de souches haploïdes. Les conditions de culture doivent être optimisées afin d’obtenir des populations
homogènes de cellules. L’expression génique est affectée par le type de milieu utilisé (même par des différences entre des lots d’un même milieu), le matériel, les conditions de température et d’aération, etc. Il convient aussi d’éviter les situations conduisant à une altération incontrôlée du niveau d’expression, telles que le passage en phase stationnaire induisant une transition à l’état diauxique, en condition de létalité cellulaire, ou encore en condition de stress métabolique lié à des changements de milieu. Les cultures comparées doivent être aussi dans le même état de croissance et si possible synchronisées au niveau du cycle cellulaire. De même, il convient d’éviter d’induire involontairement des stress métaboliques et autres
effets indésirables (choc thermique, dégradation des ARN…) lors de la préparation des échantillons. Les cellules doivent être prélevées le plus rapidement possible, sans étapes de lavage superflues, et devraient être congelées immédiatement dans de l’azote liquide.

46
V. Guide de diagnostic des anomalies
Ce guide qui illustre les problèmes techniques les plus fréquemment rencontrés. Toutes les images qui y sont présentées sont issues de mes travaux réalisés pendant les premières années de ma thèse, et j’ai listé des propositions de solutions que nous avons nous-mêmes appliquées avec succès ou qui résument des observations provenant d’autres laboratoires.
Figure 14 : Image d’une puce contenant l’ensemble des ORFs de la levure, telle que nous les avons produites au laboratoire courant 2003.
A gauche : taille réelle. Dans le cas idéal, la lame présente des spots réguliers avec une bonne amplitude du rapport signal / bruit de fond et pas d’artéfact visible à l’œil.

47
1.1. Problèmes liés à l’aspect des spots
a b c d e f Figure 15 : Photos de défauts de spotting
(a) spots réguliers ; (b) spots en anneaux ; (c) spots irréguliers dus à un mauvais calibrage du robot ; (d) spots trop petits ; (e) spots gonflés dus à une mauvaise purification des produits de PCR ; (f) particules rouges dans les spots indiquant une surface de polylysine abîmée.
Spots en anneaux (« Doughnuts »):
⇒ La solution d’ADN tend à se concentrer sur les bords du spot en séchant. L’aspect des spots peut être amélioré en ajustant le tampon d’impression (ajout éventuel de DMSO), le calibrage des pointes, l’humidité ambiante pendant impression et en réhydratant soigneusement les lames pendant le traitement de finition.
Spots déformés et irréguliers :
⇒ Pointes du spotteur tordues, bouchées, ou entraînant de la poussière. ⇒ Le spotteur frappe trop fort ou trop vite sur la surface : revoir le calibrage et le placement des pointes sur leur portoir. Les pointes fournies par le fabricant ne sont pas toujours de la même taille et certaines sont plus performantes à un emplacement qu’à un autre. Elles sont aussi parfois cassées à la réception. Des problèmes peuvent aussi provenir de la qualité du robot en soi et il peut être impossible d’y apporter une amélioration sans changer d’équipement (nous avons constaté une nette amélioration de en utilisant un robot Biorobotics plutôt que l’ancien robot Genemachines).
Spots trop petits :
⇒ Pointes trop fines. Biorobotics propose deux types de pointes utilisables avec le spotteur Omnigrid II : les « standard » (MicroSpot 2500, 100 µm de diamètre) et les « fines » (Microspot 10K, 50 µm de diamètre). Nous préférons utiliser les pointes « standard » dans nos conditions d’impression et de lecture des puces. ⇒ Pointes neuves : les pointes doivent s’user un peu pour que les spots atteignent une taille stable. ⇒ Problème de purification des produits de PCR : nous avons remarqué que la méthode de purification influe sur la qualité de l’impression. Notamment, les produits de PCR issus de plaques purifiées avec un système de filtre Millipore donnent des spots plus petits que les plaques purifiées par précipitation à l’éthanol.

48
Spots trop gros, gonflés, qui se chevauchent : ⇒ Volume de produits de PCR trop élevé dans les plaques d’impression à 384 puits. ⇒ Mauvais calibrage des pointes (force de frappe, vitesse d’impression…) ⇒ Humidité ambiante relative trop élevée (ne pas dépasser 60%) ⇒ Mauvaise purification des produits de PCR : les spots se dilatent s’il reste par exemple des traces d’éthanol (ceci arrive fréquemment avec des systèmes de purification de type QIAquick PCR purification kit).
Absence de certains spots :
⇒ Absence de produits de PCR. ⇒ Pointes bouchées.
Signal faible sur certains spots (qu’on attend fort) : ⇒ Trop faible concentration des produits de PCR.
Traces rouges dans chaque spot : ⇒ Les pointes arrachent le revêtement de polylysine pendant l’impression. Régler la force de frappe ou utiliser des lames à surface autre que la polylysine.
1.2. Problèmes de bruits de fond sur la lame
Bruit de fond vert sur une partie de la lame (souvent sur un coté) : ⇒ Mauvaise manipulation au cours du traitement avant hybridation et/ou du lavage. Le plus souvent, il s’agit du SDS qui précipite. Le SDS doit être bien rincé aux étapes de préhybridation et de lavages. Veiller à effectuer les opérations très rapidement, sans laisser les lames s’assécher entre les trempages et avant la centrifugation. Vérifier aussi la qualité des solutions utilisées (en particulier le méthyl-pyrrolidone et l’isopropanol qui doivent être limpides dans leurs flacons). Veiller enfin à ce que les chambres d’hybridation soient fermées hermétiquement.
Bruit de fond sur l’ensemble de la lame, surface peu homogène, et/ou revêtement arraché :
⇒ Problème de dégradation des lames portant un revêtement de polylysine. Très fréquent après 1 mois de conservation. Ce problème n’est pas rencontré avec les lames Corning.
Bruit de fond rouille : ⇒ Omission de l’étape de préhybridation à la BSA lors de l’utilisation de lames à surface amino-silanes. Cette étape est obligatoire avec les lames Corning Gaps II.

49
Bruits de fond blanc poussiéreux : ⇒ Poussière : Stocker les lames dans une boîte propre, ne pas utiliser de gants poudrés... ⇒ Précipitation de sels : vérifier la composition du tampon d’hybridation de la cible et veiller à ce que les chambres d’hybridation soient hermétiquement fermées.
« Comètes », important bruit de fond autour des spots ⇒ Concentration trop élevée des produits de PCR à déposer. La concentration doit être comprise entre 100-250 µg/ml ⇒ Mauvais traitement de finition des lames (mauvais blocage à l’anhydride succinique) ⇒ Lot de lames défectueux (peut notamment indiquer une mauvaise élimination de la soude NaOH pendant la préparation des lames de polylysine).
a b c
d e f Figure 16 : Photos de lames avec problèmes de bruits de fond et de signal
(a) trace de SDS mal éliminé ; (b) lame Corning traitée sans préhybridation à la BSA ; (c) revêtement de polylysine dégradé ; (d) cristallisation de sels ; (e) « comètes » ; (f) signal plus faible au centre de la lame à cause d’une lamelle d’hybridation non plane.
1.3. Faible intensité de signal
Rapport bruit de fond/signal faible ⇒ Problèmes au niveau du marquage de la cible. Parmi les causes les plus couramment rencontrées nous pouvons citer : - Une quantité de matériel de départ trop faible : vérifier la concentration et la qualité
des ARN messagers utilisés.

50
- Des esters de fluorochromes dégradés : ils doivent être conservés sous forme de culots secs et ne jamais entrer en contact avec de l’eau avant utilisation.
- Un oubli de l’ajout de bicarbonate de sodium avant l’étape de couplage avec les colorants.
Signal faible sur une partie de la lame (souvent au centre), hybridation non homogène :
⇒ Les lamelles couvrantes ne sont pas planes. Nous avons utilisé pour nos projets des lamelles en plastique souples qui sont commodes pour diverses raisons (facilité de manipulation, surface propre, peu de risque de formation de bulles…) mais ne sont pas idéales à cet égard. Il peut être intéressant de choisir des lamelles surélevées.
1.4. Données biaisées détectées à l’analyse
Valeurs aberrantes à un des points d’une cinétique (par exemple, diminution brutale de l’expression à un point seulement alors que l’expression semble augmenter régulièrement de manière constante au cours de la cinétique) :
⇒ Cas simple : erreur de permutation de deux points de la cinétique lors de l’analyse. ⇒ Cas complexe : erreur de permutation entre deux échantillons biologiques pendant une étape de la manipulation expérimentale. ⇒ Cas insoluble : problème de dégradation de l’ARN de départ. Si l’ARN est dégradé, on peut obtenir des valeurs totalement aberrantes car la dégradation ne se fait pas de manière linéaire.
Expression particulièrement faible aux premiers temps d’une série chronologique : ⇒ Choix inapproprié de l’échantillon de référence. Par exemple, dans une étude du programme transcriptionnel de la méiose, si les cellules commencent à entrer en méiose avant le temps initial t=0h choisi comme référence, les gènes exprimés aux étapes précoces de la méiose n’apparaissent pas induits relativement a cette référence car le niveau des transcrits est déjà élevé à t=0h.

51
VI. Protocoles expérimentaux
Protocole 1 : Amplification par PCR et purification des gènes-cibles à déposer.
Les amorces Research Genetics sont conservées à –80 °C (dans des plaques 96 puits). Décongeler une nuit à 4 °C (essuyer les plaques pour que de l’eau ne s’infiltre pas sous les couvercles). Transférer 5 µl de chaque amorce dans les plaques à PCR à l’aide d’un robot pipeteur. Important : bien mélanger chaque solution d’amorces dans les plaques pour homogénéiser avant utilisation. Préparer un mélange de réaction de 40 ml (volume correspondant à 4 plaques PCR, 100 µl par puits) :
Tampon 10x 4000 µl MgCl2 (25 mM) 3200 µl dNTP mix 400 µl (dATP, dCTP, dTTP, dGTP 2,5 mM chacun) Taq polymérase (1: 40) 15 µl ADN génomique (1 µg/µl) 20 µl H2O q.s.p. 36000 µl
Distribuer 90µl par puits de PCR. Couvrir avec un tapis de caoutchouc, lancer le programme de PCR sur un thermocycler : 36 cycles : 92 °C 30s ==> 52 °C 45s ==> 72 °C 3mn30 Vérifier la taille des produits en déposant 2µl sur un gel d’électrophorèse Ajouter à l’aide d’un robot dans chaque puits : 10 µl sodium acétate 3 M + 110 µl isopropanol frais Précipiter o/n minimum à –20 °C Centrifuger 4000 rpm 2h30 (sur une centrifugeuse Sorvall RC3B+) Rincer avec 75 µl d’éthanol 70% Centrifuger 4000 rpm 30mn Aspirer le surnageant et sécher au speed-vacuum Reprendre dans 55 µl H2O et laisser se dissoudre 24h minimum a 4 °C Aliquoter à l’aide d’un robot 10µl par plaque de spotting 384-puits Sécher au speed-vacuum et conserver à –20°C Avant le dépôt sur lames de verre : ajouter 5 µl de 3x SSC, laisser se re-dissoudre à 4°C, 24h minimum.
Protocole 2 : Traitement des lames avant hybridation.
Numéroter et marquer les lames avec un graveur pour verre pour repérer les spots (qui deviennent invisibles après le traitement par dissolution des sels du tampon de dépôt). Réhydrater les spots à la vapeur en retournant les lames au dessus d’un réservoir d’eau chaude à 50 °C pendant environ 10s. Sécher 3s en les posant sur une plaque chauffante à environ 80 °C. Traiter à l’irradiation dans un Stratalinker UV 260 mJ. Préparer la solution de blocage (à préparer très rapidement juste avant le traitement) :
anhydride succinique 3 g méthylpyrrolidone 150 ml
+ borate de sodium 0,2 M pH=8 17 ml dès dissolution de l’anhydride

52
Remplir une cuve en verre de la solution et y plonger les lames, couvrir et incuber 20mn avec une faible agitation sur un plateau à bascule. Dénaturer en les plongeant dans un bêcher de 2-3 L d’eau froide puis dans de l’éthanol 95% préalablement refroidi à –20 °C. Centrifuger 5mn à environ 50 g à température ambiante pour sécher. Attention, la centrifuge ne doit surtout pas être froide pour éviter la formation de condensation. Préparer la solution de préhybridation (pour 5 lames) :
H2O 24,45 ml 20X SSC 5,25 ml 10% SDS 300 µl BSA 0,3 g
Filtrer sur un filtre à seringue millipore 0,.22 µm et verser dans un étui à lames Corning pour préchauffer 10mn à 50 °C dans un four. Incuber les lames 30mn à 50 °C. Rincer 1mn dans H20 et 1mn dans de l’isopropanol à température ambiante. Centrifuger 5mn à environ 50 g à température ambiante pour sécher.
Protocole 3 : Extraction des ARN totaux au phénolchloroforme à chaud avec les tubes Phase-lock Gel Eppendorf
Re-suspendre rapidement les cellules congelées dans 1,5ml de tampon TES (Tris 10 mM pH=7,5 ; EDTA 10 mM ; SDS 0,5%) et transvaser dans un tube falcon 15 ml. Ajouter 1 ml phénolchloroforme 5:1 préchauffé à 65 °C (phénol acide équilibré avec 20 mM NaAc pH=5,2) et 500 µg de billes de verre lavées à l’acide (optionnelles pour les cellules végétatives, mais nécessaires pour les cellules en sporulation). Bien mélanger en vortexant. Incuber 1h à 65 °C en vortexant 20s toutes les 10mn. Refroidir 5mn dans la glace. Centrifuger à 4 °C, 1500 g, 5mn. Centrifuger en même temps un tube Phase-lock Gel Eppendorf15ml par échantillon. Verser la phase aqueuse dans le tube Phase-lock Gel. Ajouter 1,5ml de phénolchloroforme, vortexer 20s. Centrifuger à 4 °C, 1500 g, 5mn. Répéter 3 fois dans le même tube. Ajouter 1,5ml de chloroforme-alcool isoamylique 25:1, vortexer 20s Centrifuger à 4 °C, 1500 g, 5mn. Transférer la phase aqueuse dans des tubes eppendorfs 2ml. Précipiter dans 2 volumes d’éthanol 95% et 1/10e de volume d’acétate de sodium 3M – DEPC pendant au moins 1/2h à –20 °C. Centrifuger à 4 °C, 13000 g, 5mn. Rincer le culot à l’éthanol 80% froid, vortexer brièvement. Centrifuger à 4 °C, 13000 g, 5mn. Sécher le culot (à l’air ou au speed-vacuum mais pas trop longtemps pour ne pas laisser durcir). Reprendre dans 100 µl H2O-DEPC et bien dissoudre. Conserver à –80 °C. Pour la vérification par électrophorèse : 1 µl d’ARN dénaturé à la chaleur (5mn à 95 °C puis immédiatement refroidi sur la glace) est fractionné sur un gel à 1% d’agarose dans un tampon TBE. Le gel est coloré pendant 10mn dans une solution de bromure d’éthidium (1 µg/ml) puis révélé sous UV.

53
Protocole 4 : Extraction des ARNm avec le kit Micro-FastTrack (Invitrogen)
Ajouter sur les ARN totaux 950 µl de « Stock Buffer » (Tampon de lyse) et 20 µl de « Protein/RNase Degrader ». Chauffer 65 °C, 5mn. Refroidir 1mn dans la glace. A température ambiante, ajouter 63 µl de « NaCl 5 M ». Ajouter la solution d'ARN aux billes d'oligo(dT) cellulose, mélanger doucement. Incuber 2h sur un portoir rotatif. Centrifuger à 4000 g pendant 5mn, enlever le surnageant. Resuspendre le culot dans 1,3 ml de « Binding Buffer » (Tampon de fixation). (x 2) Centrifuger à 4000 g pendant 5mn, enlever le surnageant. Resuspendre dans 0,3 ml de « Binding Buffer », transférer dans une « Spin-column ». Centrifuger à 4000 g pendant 10 s, jeter l’effluent. Ajouter 0,5 ml de « Binding Buffer ». Centrifuger à 4000 g pendant 10 s, jeter l’effluent. Ajouter 200 µL de « Low Salt Wash Buffer » (Tampon de lavage), resuspendre avec une pointe de pipette (Essayer de désagréger les plus gros amas de billes avec la pointe, éviter de toucher la membrane). (x 2) Centrifuger à 4000 g pendant 10 s , jeter l’effluent. Placer la colonne sur un nouveau tube stérile. Ajouter 200 µl « Elution Buffer » (Tampon d’élution), resuspendre avec une pointe de pipette. (x 2) Centrifuger à 4000 g pendant 5mn Récupérer l’effluent, précipiter les ARNm dans 800 µl d’éthanol 95% et 60 µl de « NaAc 2 M » pendant une nuit à –80 °C. Centrifuger à 16000 g, 30mn, 4 °C. Rincer à l'éthanol 80 %. Centrifuger à 16000 g, 15mn, 4 °C. Sécher et reprendre dans 10 µl d’H2O-DEPC, stocker à –80 °C.
Protocole 5 : Synthèse des cibles fluorescentes par couplage indirect à l’amino-allyl dUTP
1. Dénaturation de l’ARN Mélanger dans un tube eppendorf 0,5 ml :
1-2 µg ARNm 5 µg amorces aléatoires d’hexamers Pd(N)6 5 µg amorces oligo dT q.s.p. 15,5 µl H2O-DEPC
Chauffer 10mn à 70 °C Refroidir immédiatement dans de la glace 2. Synthèse de l’ADNc par la transcription inverse Ajouter : 6 µl tampon 5x 3 µl DTT 0,1 M 0,6 µl mix dNTP 50x (dATP, dCTP, dGTP 25 mM, aa-dUTP 15 mM, dTTP 10 mM) 3 µl H2O-DEPC 2 µl Transcriptase Reverse SuperScript II 200 U/µl Incuber 2h à 42 °C

54
3. Hydrolyse de l’ARN Ajouter : 10 µl NaOH 1 N 10 µl EDTA 0,5 M Incuber 15mn à 65 °C Ajouter : 25 µl TRIS 1 M pH = 7,4 (neutralisation) 4. Rinçage Mettre 450 µl H2O dans une colonne microcon-30 (Millipore) et ajouter l’ADNc Centrifuger 7mn à 10000 g à température ambiante, jeter l’effluent. Répéter 2 fois dans la même colonne (ajouter 450 µl H2O et centrifuger) La dernière centrifugation doit être ajustée en temps de manière à ce qu’il reste moins de 10 µl de volume final (ajuster « à vue d’œil ») Centrifuger 2mn à 1000 g en retournant la colonne pour récupérer l’échantillon d’ADNc L’ADNc peut être conservé indéfiniment à –20 °C 5. Préparer des aliquotes de fluorochromes : Le NHS-ester de Cy3 ou Cy5 distribué par Amersham est fourni sous forme lyophilisée. Re-dissoudre un tube de NHS-ester de Cy3/Cy5 dans 10 µl de DMSO anhydre (éviter la contamination par des solutions aqueuses car les fluorochromes perdent très vite leur réactivité en présence d’eau). Aliquoter 1,25 µl x 8 tubes et sécher immédiatement au speed-vacuum pour conserver au sec à 4 °C (ou utiliser immédiatement sans sécher) 6. Couplage du NHS-ester Cy3/Cy5 Ajuster l’échantillon d’ADNc à 10 µl avec H2O. Ajouter 0,5 µl de Bicarbonate de Sodium 1M pH = 9 pour obtenir une solution de concentration finale 0,05 M Ajouter l’échantillon d’ADNc à une aliquote d’esters de Cy3 ou Cy5, bien mélanger à la pipette. Incuber 1h à température ambiante dans l’obscurité 7. « Quenching » Ajouter 4,5 µl d’Hydroxylamine 4 M Incuber 15mn à température ambiante dans l’obscurité NB : le quenching est supposé arrêter la réaction et empêcher les interactions indésirables entre fluorochomes et nucléotides, mais certains protocoles récents indiquent que cette étape peut être omise. 8. Purification Mélanger les deux cibles marqués au Cy3 et au Cy5, ajouter 70 µl H2O
Purifier avec une colonne QiaQuick PCR Purification kit (Qiagen) : Ajouter 500 µl de tampon PB, bien mélanger et placer la solution sur la colonne Centrifuger 30s 13000 g , jeter le flow-through Ajouter 750 µl de tampon de lavage PE (préparé avec de l’éthanol 95% non fluorescent) Centrifuger 30s, jeter l’effluent, répéter Centrifuger à vide 1mn pour retirer toute trace de tampon PE Eluer sur un nouveau tube eppendorf avec 30 µl de tampon EB Centrifuger 1mn, répéter
Concentrer 3mn dans une colonne microcon-30 Centrifuger 2mn à 1000 g en retournant la colonne pour récupérer la cible marquée qui doit être vivement colorée en violet

55
Protocole 6 : Hybridation de la puce à ADN
Ajuster la cible marquée d’ADNc préparée dans le protocole précédent à 30 µl avec H2O Ajouter : 6 µl SSC 20x 3 µl poly A (10 µg/µl) 1 µl HEPES 1 M pH = 7,0 La cible peut être conservée 24h à 4°C Ajouter : 0,9 µl de SDS 10% Dénaturer 2mn à 95 °C et laisser refroidir à température ambiante (ne pas refroidir dans la glace pour ne pas faire précipiter le SDS) Déposer 15 µl de SSC 3x dans les creux réservés au fond de la chambre d’hybridation Placer une lame de microarray, spots sur le dessus Déposer la cible à l’emplacement des spots Recouvrir d’une lamelle souple (en évitant les bulles au maximum) Déposer une goutte de SSC 3x sur les quatre coins de la lame (sans toucher la lamelle) Fermer la chambre (hermétiquement, mais ne pas trop serrer les vis) Incuber dans un bain-marie à 63 °C pendant une nuit (minimum 8h)
Protocole 7 : Lavage de la puce à ADN
Préparer les solutions de lavage : A : 190 ml H2O B : 198 ml H2O C: 200 ml H2O 10 ml SSC 20x 2 ml SSC 20x 0,5 ml SSC 20x 600 µl SDS 10% Retirer la lame de la chambre d’hybridation, la placer sur un portoir métallique pour la placer immédiatement dans une cuve en verre contenant la solution A La lamelle se décolle en quelques secondes. La laisser tomber au fond. Placer rapidement la lame sur un autre portoir propre (pour ne pas emporter trop de SDS) et plonger plusieurs fois dans la solution B Laisser tremper 3-5mn à faible agitation. Répéter avec la solution C en changeant de portoir Attention : ne pas laisser la lame sécher entre chaque transfert Sécher en centrifugeant 3mn à 50 g à température ambiante Ranger dans une boite en plastique à l’abri des poussières et de l’humidité, scanner dans la journée

56
VII. Références bibliographiques
1. Sollier, J., et al., Set1 is required for S phase onset, double-strand break formation and middle gene expression during meiosis in Saccharomyces cerevisiae. Embo J, 2004. 23: p. 1957-67.
2. Borde, V., et al., Association of Mre11p with double-strand break sites during yeast meiosis. Mol Cell, 2004. 13(3): p. 389-401.
3. DNA Microarrays. A Molecular Cloning Manual, ed. D. Bowtell and J. Sambrook. 2003, New York: Cold Spring Harbor Laboratory Press. 712.
4. The Chipping Forecast. Nat Genet, 2002. 32 Suppl: p. 461-552.
5. Brent, R., Genomic biology. Cell, 2000. 100(1): p. 169-83.
6. Benson, D.A., et al., GenBank: update. Nucleic Acids Res, 2004. 32 Database issue: p. D23-6.
7. Lander, E.S., et al., Initial sequencing and analysis of the human genome. Nature, 2001. 409(6822): p. 860-921.
8. Venter, J.C., et al., The sequence of the human genome. Science, 2001. 291(5507): p. 1304-51.
9. Goffeau, A., et al., Life with 6000 genes. Science, 1996. 274(5287): p. 546, 563-7.
10. Cherry, J.M., et al., SGD: Saccharomyces Genome Database. Nucleic Acids Res, 1998. 26(1): p. 73-9.
11. Mewes, H.W., et al., MIPS: a database for protein sequences, homology data and yeast genome information. Nucleic Acids Res, 1997. 25(1): p. 28-30.
12. Hodges, P.E., W.E. Payne, and J.I. Garrels, The Yeast Protein Database (YPD): a curated proteome database for Saccharomyces cerevisiae. Nucleic Acids Res, 1998. 26(1): p. 68-72.
13. Velculescu, V.E., et al., Characterization of the yeast transcriptome. Cell, 1997. 88(2): p. 243-51.
14. Kumar, A. and M. Snyder, Emerging technologies in yeast genomics. Nat Rev Genet, 2001. 2(4): p. 302-12.
15. Grunenfelder, B. and E.A. Winzeler, Treasures and traps in genome-wide data sets: case examples from yeast. Nat Rev Genet, 2002. 3(9): p. 653-61.
16. Ross-Macdonald, P., et al., Large-scale analysis of the yeast genome by transposon tagging and gene disruption. Nature, 1999. 402(6760): p. 413-8.
17. Winzeler, E.A., et al., Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science, 1999. 285(5429): p. 901-6.
18. Giaever, G., et al., Functional profiling of the Saccharomyces cerevisiae genome. Nature, 2002. 418(6896): p. 387-91.
19. Huh, W.K., et al., Global analysis of protein localization in budding yeast. Nature, 2003. 425(6959): p. 686-91.
20. Ghaemmaghami, S., et al., Global analysis of protein expression in yeast. Nature, 2003. 425(6959): p. 737-41.

57
21. DeRisi, J.L., V.R. Iyer, and P.O. Brown, Exploring the metabolic and genetic control of gene expression on a genomic scale. Science, 1997. 278(5338): p. 680-6.
22. Wodicka, L., et al., Genome-wide expression monitoring in Saccharomyces cerevisiae. Nat Biotechnol, 1997. 15(13): p. 1359-67.
23. Zhu, H. and M. Snyder, Protein chip technology. Curr Opin Chem Biol, 2003. 7(1): p. 55-63.
24. Uetz, P., et al., A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature, 2000. 403(6770): p. 623-7.
25. Ito, T., et al., A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci U S A, 2001. 98(8): p. 4569-74.
26. Ren, B., et al., Genome-wide location and function of DNA binding proteins. Science, 2000. 290(5500): p. 2306-9.
27. Iyer, V.R., et al., Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature, 2001. 409(6819): p. 533-8.
28. Martzen, M.R., et al., A biochemical genomics approach for identifying genes by the activity of their products. Science, 1999. 286(5442): p. 1153-5.
29. Ho, Y., et al., Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature, 2002. 415(6868): p. 180-3.
30. Gavin, A.C., et al., Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature, 2002. 415(6868): p. 141-7.
31. Southern, E.M., Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol, 1975. 98(3): p. 503-17.
32. Lipshutz, R.J., et al., High density synthetic oligonucleotide arrays. Nat Genet, 1999. 21(1 Suppl): p. 20-4.
33. Hughes, T.R., et al., Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat Biotechnol, 2001. 19(4): p. 342-7.
34. Holloway, A.J., et al., Options available--from start to finish--for obtaining data from DNA microarrays II. Nat Genet, 2002. 32 Suppl: p. 481-9.
35. Spellman, P.T., et al., Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell, 1998. 9(12): p. 3273-97.
36. Cho, R.J., et al., A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell, 1998. 2(1): p. 65-73.
37. Chu, S., et al., The transcriptional program of sporulation in budding yeast. Science, 1998. 282(5389): p. 699-705.
38. Primig, M., et al., The core meiotic transcriptome in budding yeasts. Nat Genet, 2000. 26(4): p. 415-23.
39. Kuhn, K.M., et al., Global and specific translational regulation in the genomic response of Saccharomyces cerevisiae to a rapid transfer from a fermentable to a nonfermentable carbon source. Mol Cell Biol, 2001. 21(3): p. 916-27.
40. Gasch, A.P., et al., Genomic expression responses to DNA-damaging agents and the regulatory role of the yeast ATR homolog Mec1p. Mol Biol Cell, 2001. 12(10): p. 2987-3003.

58
41. Hughes, T.R., et al., Functional discovery via a compendium of expression profiles. Cell, 2000. 102(1): p. 109-26.
42. Sorlie, T., et al., Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A, 2001. 98(19): p. 10869-74.
43. Alizadeh, A.A., et al., Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature, 2000. 403(6769): p. 503-11.
44. Wang, Y., et al., Precision and functional specificity in mRNA decay. Proc Natl Acad Sci U S A, 2002. 99(9): p. 5860-5.
45. Diehn, M., et al., Large-scale identification of secreted and membrane-associated gene products using DNA microarrays. Nat Genet, 2000. 25(1): p. 58-62.
46. Marc, P., et al., Genome-wide analysis of mRNAs targeted to yeast mitochondria. EMBO Rep, 2002. 3(2): p. 159-64.
47. Chi, J.T., et al., Genomewide view of gene silencing by small interfering RNAs. Proc Natl Acad Sci U S A, 2003. 100(11): p. 6343-6.
48. Jackson, A.L., et al., Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol, 2003. 21(6): p. 635-7.
49. Arava, Y., et al., Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A, 2003. 100(7): p. 3889-94.
50. Clark, T.A., C.W. Sugnet, and M. Ares, Jr., Genomewide analysis of mRNA processing in yeast using splicing-specific microarrays. Science, 2002. 296(5569): p. 907-10.
51. Peng, W.T., et al., A panoramic view of yeast noncoding RNA processing. Cell, 2003. 113(7): p. 919-33.
52. Simon, I., et al., Serial regulation of transcriptional regulators in the yeast cell cycle. Cell, 2001. 106(6): p. 697-708.
53. Lieb, J.D., et al., Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat Genet, 2001. 28(4): p. 327-34.
54. Lee, T.I., et al., Transcriptional regulatory networks in Saccharomyces cerevisiae. Science, 2002. 298(5594): p. 799-804.
55. Kurdistani, S.K., et al., Genome-wide binding map of the histone deacetylase Rpd3 in yeast. Nat Genet, 2002. 31(3): p. 248-54.
56. Ng, H.H., et al., Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell, 2003. 11(3): p. 709-19.
57. Damelin, M., et al., The genome-wide localization of Rsc9, a component of the RSC chromatin-remodeling complex, changes in response to stress. Mol Cell, 2002. 9(3): p. 563-73.
58. Ng, H.H., et al., Genome-wide location and regulated recruitment of the RSC nucleosome-remodeling complex. Genes Dev, 2002. 16(7): p. 806-19.
59. Robyr, D., et al., Microarray deacetylation maps determine genome-wide functions for yeast histone deacetylases. Cell, 2002. 109(4): p. 437-46.
60. Huang, T.H., M.R. Perry, and D.E. Laux, Methylation profiling of CpG islands in human breast cancer cells. Hum Mol Genet, 1999. 8(3): p. 459-70.

59
61. Wyrick, J.J., et al., Genome-wide distribution of ORC and MCM proteins in S. cerevisiae: high-resolution mapping of replication origins. Science, 2001. 294(5550): p. 2357-60.
62. Gerton, J.L., et al., Inaugural article: global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci U S A, 2000. 97(21): p. 11383-90.
63. Keeney, S., C.N. Giroux, and N. Kleckner, Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell, 1997. 88(3): p. 375-84.
64. Kallioniemi, A., et al., Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science, 1992. 258(5083): p. 818-21.
65. Pollack, J.R., et al., Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet, 1999. 23(1): p. 41-6.
66. Pinkel, D., et al., High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet, 1998. 20(2): p. 207-11.
67. Raghuraman, M.K., et al., Replication dynamics of the yeast genome. Science, 2001. 294(5540): p. 115-21.
68. Dunham, M.J., et al., Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A, 2002. 99(25): p. 16144-9.
69. Wang, D.G., et al., Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science, 1998. 280(5366): p. 1077-82.
70. Winzeler, E.A., et al., Direct allelic variation scanning of the yeast genome. Science, 1998. 281(5380): p. 1194-7.
71. Birrell, G.W., et al., A genome-wide screen in Saccharomyces cerevisiae for genes affecting UV radiation sensitivity. Proc Natl Acad Sci U S A, 2001. 98(22): p. 12608-13.
72. Birrell, G.W., et al., Transcriptional response of Saccharomyces cerevisiae to DNA-damaging agents does not identify the genes that protect against these agents. Proc Natl Acad Sci U S A, 2002. 99(13): p. 8778-83.
73. Deutschbauer, A.M., et al., Parallel phenotypic analysis of sporulation and postgermination growth in Saccharomycescerevisiae. Proc Natl Acad Sci U S A, 2002. 99(24): p. 15530-5.
74. Ooi, S.L., D.D. Shoemaker, and J.D. Boeke, A DNA microarray-based genetic screen for nonhomologous end-joining mutants in Saccharomyces cerevisiae. Science, 2001. 294(5551): p. 2552-6.
75. Ooi, S.L., D.D. Shoemaker, and J.D. Boeke, DNA helicase gene interaction network defined using synthetic lethality analyzed by microarray. Nat Genet, 2003. 35(3): p. 277-86.
76. van de Peppel, J., et al., Monitoring global messenger RNA changes in externally controlled microarray experiments. EMBO Rep, 2003. 4(4): p. 387-93.
77. Randolph, J.B. and A.S. Waggoner, Stability, specificity and fluorescence brightness of multiply-labeled fluorescent DNA probes. Nucleic Acids Res, 1997. 25(14): p. 2923-9.
78. Yang, Y.H., et al., Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res, 2002. 30(4): p. e15.
79. Saal, L.H., et al., BioArray Software Environment (BASE): a platform for comprehensive management and analysis of microarray data. Genome Biol, 2002. 3(8): p. SOFTWARE0003.
80. Sturn, A., Cluster Analysis for Large Scale Gene Expression Studies. 2000, The Institute for Genomic Research: Rockville, Maryland, USA,. p. 71.

60
81. Ge, H., et al., Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat Genet, 2001. 29(4): p. 482-6.
82. Jansen, R., D. Greenbaum, and M. Gerstein, Relating whole-genome expression data with protein-protein interactions. Genome Res, 2002. 12(1): p. 37-46.
83. Eisen, M.B., et al., Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A, 1998. 95(25): p. 14863-8.
84. Sturn, A., J. Quackenbush, and Z. Trajanoski, Genesis: cluster analysis of microarray data. Bioinformatics, 2002. 18(1): p. 207-8.
85. Ashburner, M., et al., Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet, 2000. 25(1): p. 25-9.
86. Dwight, S.S., et al., Saccharomyces Genome Database (SGD) provides secondary gene annotation using the Gene Ontology (GO). Nucleic Acids Res, 2002. 30(1): p. 69-72.
87. Kellis, M., et al., Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature, 2003. 423(6937): p. 241-54.
88. Cliften, P., et al., Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science, 2003. 301(5629): p. 71-6.
89. Brachat, S., et al., Reinvestigation of the Saccharomyces cerevisiae genome annotation by comparison to the genome of a related fungus: Ashbya gossypii. Genome Biol, 2003. 4(7): p. R45.
90. Kumar, A., et al., An integrated approach for finding overlooked genes in yeast. Nat Biotechnol, 2002. 20(1): p. 58-63.
91. Gaasterland, T. and M. Oprea, Whole-genome analysis: annotations and updates. Curr Opin Struct Biol, 2001. 11(3): p. 377-81.
92. Wiederkehr, C., et al., GermOnline, a cross-species community knowledgebase on germ cell differentiation. Nucleic Acids Res, 2004. 32 Database issue: p. D560-7.
93. Marc, P., F. Devaux, and C. Jacq, yMGV: a database for visualization and data mining of published genome-wide yeast expression data. Nucleic Acids Res, 2001. 29(13): p. E63-3.
94. Brazma, A., et al., ArrayExpress--a public repository for microarray gene expression data at the EBI. Nucleic Acids Res, 2003. 31(1): p. 68-71.
95. Yang, Y.H. and T. Speed, Design issues for cDNA microarray experiments. Nat Rev Genet, 2002. 3(8): p. 579-88.





























