Embed Size (px)
Citation preview

Stage de Pré-Rentrée UE 1
Organisation du stage de pré-rentrée UE 1- 9 h de chimie : Chimie générale (Atomes et liaisons), Chimie organique- 9 h de biochimie : Protides, Glucides, Lipides, Ac. nucléiques, Enzymologie
Des QCMs sont fournis avec le poly → sont à faire et seront corrigés la séance suivante
Dans un chapitre, des questions simples « participatives » et récapitulatives à chaque fin de partie → Donc on écoute bien, on se concentre et surtout ON PARTICIPE !
Bon courage et vive la chimie !

Stage de Pré-Rentrée 2012UE 1 – L’atome
2
Diaporama réalisé par les tuteurs de La Fed’

Plan du cours
I- Structure de l’atome
II- Modèle de Bohr
III- Modèle quantique de l’atome
IV- Configuration électronique
V- Tableau périodique
I- Structure de l’atome
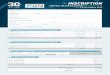
Généralités
Atome : électriquement neutre
Noyau : nucléons = neutrons + protonsconcentre la masse de l’atomeprotons : charge positive
Nuage électronique : électronscharge négative
I- Structure de l’atome
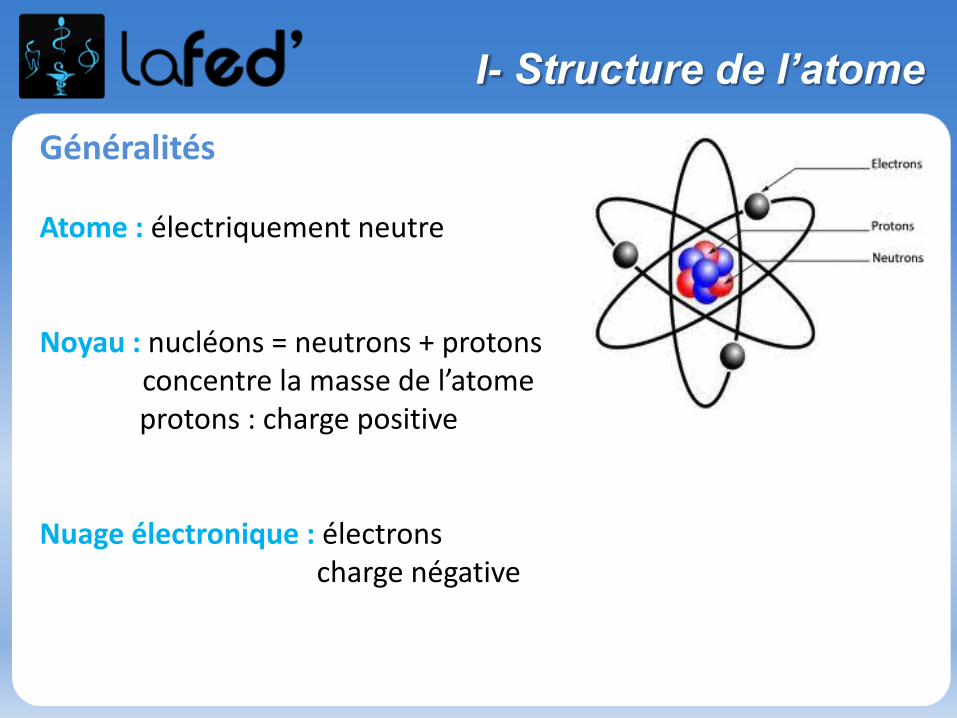
Particules élémentaires
Charge Masse
Neutron 0 C 1,672.10-27 kg
Proton + 1,602.10-19 C 1,672.10-27 kg= 1836 melectron
Electron - 1,602.10-19 C 9,109.10-31 kg

I- Structure de l’atome
Caractérisation de l’atome
A = nombre de masse
= nombre de nucléons
Z = numéro atomique
= nombre de protons
(= nombre d’électrons si pas d’ionisation
car atome électriquement neutre)
N = A – Z = nombre de neutrons
Pour les éléments légers : en général, Z = N

I- Structure de l’atome
Isotopes
Deux atomes sont des isotopes si : même Zdifférents A
Même nombre d’électrons : « isoélectroniques »
Ex: Hydrogène (p=1; n=0)Deutérium (p=1; n=1) isotope stableTritium (p=1; n=2) isotope radioactif
Carbone 12 (p=6; n=6)Carbone 13 (p=6; n=7) isotope stableCarbone 14 (p=6; n=8) isotope radioactif

I- Structure de l’atome
La mole Abréviation : mol= quantité de matière d’un système contenant autant
d’entités qu’il y a d’atomes dans 12 g de 12C.
La masse d’une mole de particules = masse molaire (unité = g.mol-1 notée M)
Le nombre d’Avogadro Abréviation : NA
= nombre d’atomes réels contenus dans un atome gramme soit dans une mole de ces entités.
NA = M/m = 6,02.1023 mol-1
Une mole de n’importe quelle substance contient donc 6,022.1023 particules de cette substance.

I- Structure de l’atome
L’unité de masse atomique Abréviation: uma
= 1/12 masse d’un atome de carbone pris à 12g.
1 uma = 1/12 x 1,9926.10-23 g = 1,6605.10-24g
Ex: Masse atomique d’un atome du nucléide 14N = 14 uma.Masse molaire d’une mole de 14N, cad la masse de 6,022.1023 atomes de 14N ≈ 14 g.mol-1.

Exercices
Au fait, une mole de grains de sable, ça fait combien de grains de sable ?!?
I- Structure de l’atome
Soit l’atome représenté ainsi :
Fe56
- Connaissez-vous le nom de cet élément ?- Quelle est sa charge globale ?- Combien de protons, d’électrons, de neutrons ?
Il existe d’autres atomes dans la nature que l’on peut écrire
- Comment peut-on qualifier sa relation avec 56Fe ?- A-t-il des propriétés chimiques différentes ? Pourquoi ?
26
Fe58
26
Rappel lycée: spectre de la lumière blanche
Lumière blanche
II- Modèle de Bohr

Relation rayonnement-énergie
La lumière peut-être considérée comme - la résultante d’une onde électromagnétique avec une certaine fréquence ν (modèle ondulatoire) - un faisceau de particules photoniques d’une certaine énergie E (modèle corpusculaire).
Ces deux modèles sont reliés par l’équation de Planck :
E = h x ν = h x c/λavec E : énergie (en Joules) NB : 1 eV = 1,6.10-19 J
c : célérité, vitesse de la lumière (3.108 m.s-1)λ : longueur d’onde (en m)h : constante de Planck (6,62.10-34 J.s-1)ν : fréquence (en s-1 ou Hz)
II- Modèle de Bohr
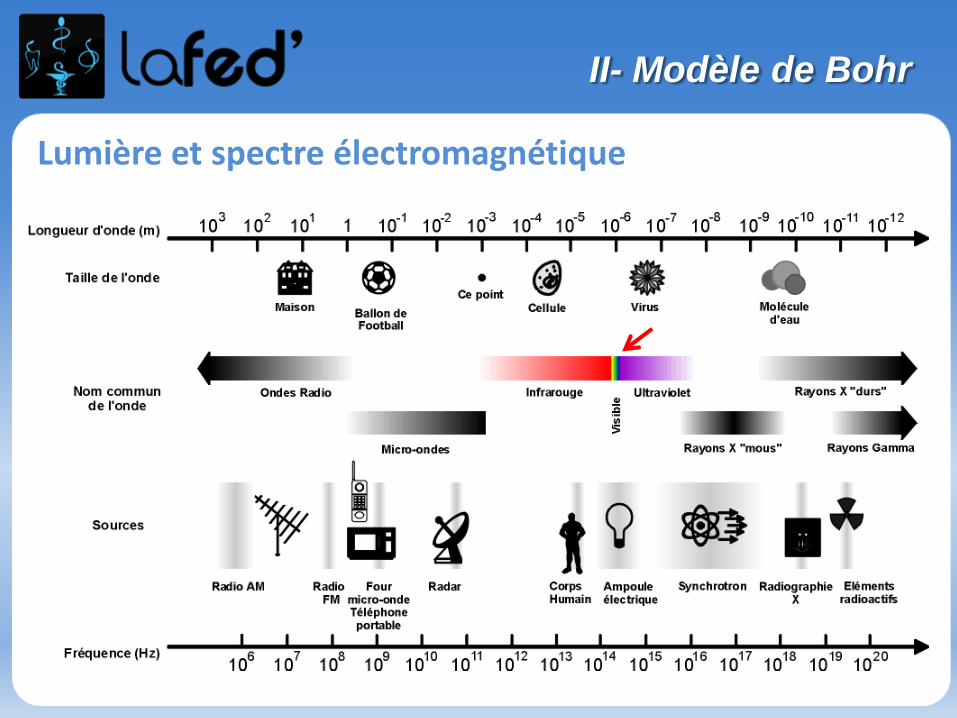
Lumière et spectre électromagnétique
II- Modèle de Bohr
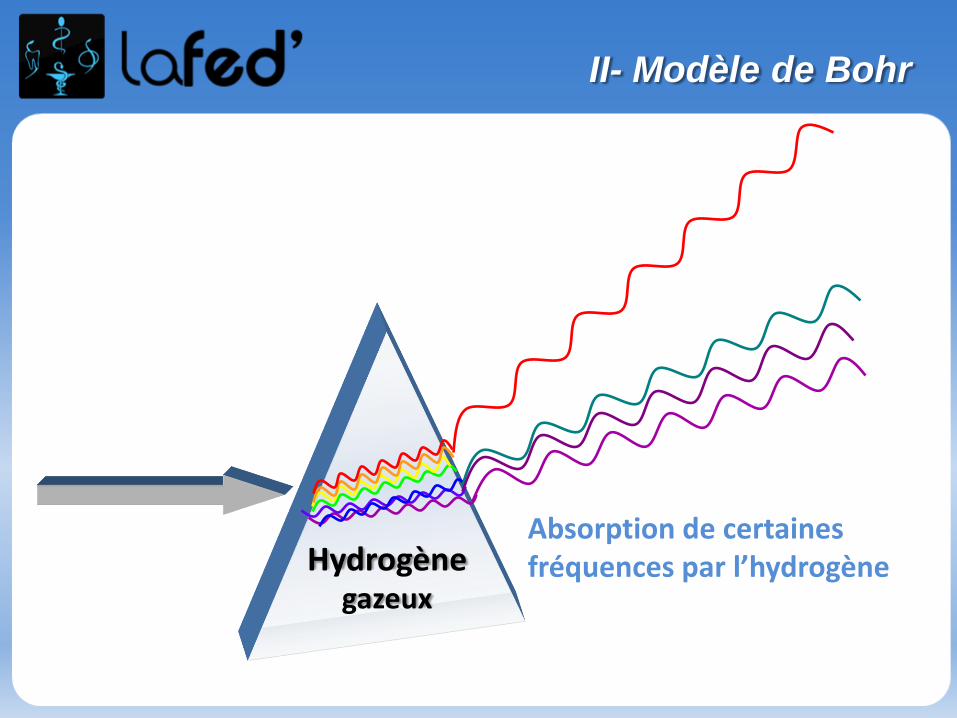
Hydrogènegazeux
Absorption de certaines fréquences par l’hydrogène
II- Modèle de Bohr

II- Modèle quantique de l’atome
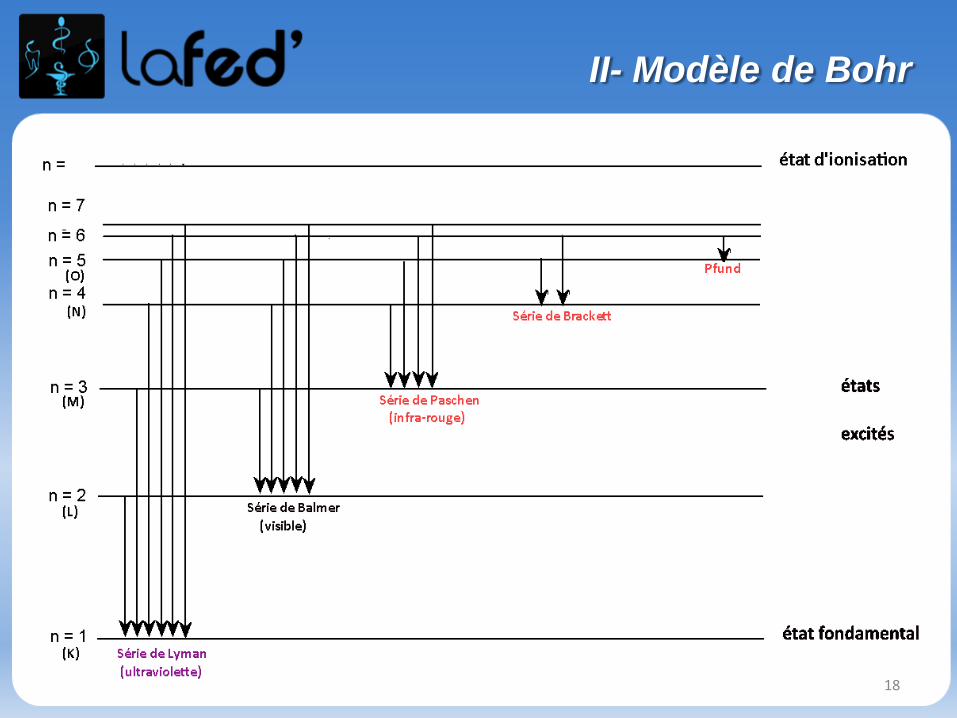
Ces spectres sont discontinus →
Le modèle de Bohr est basé surl’interprétation des spectres deraie d’absorption et d’émissionde l’atome d’hydrogène.
• Si on laisse les atomes d’hydrogènes se « désexciter », ilsémettront par fluorescence un spectre de raies d’émission quisera l’opposé du spectre d’absorption précédent.
• L’absorption de certaines fréquences particulières parl’hydrogène gazeux produit un spectre de raies d’absorption .

Toujours fondé sur les lois de la mécanique classique il inclut néanmoins certainsconcepts tels que la quantification des niveaux d’énergie (Max Planck,mécanique quantique).
Même si ce modèle est aujourd’hui considéré comme faux, il reste encore trèsintéressant, puisqu’il conduit à des résultats exacts dans certains champsd’application.
Il commence à envisager la dualité onde-particule : l’électron est à la foisconsidéré comme une particule (avec une masse) et comme une onde defréquence définie.
II- Modèle de Bohr
Modèle de BOHR
Le modèle de Niels Bohr (1885-1962) reprend les bases du modèle planétaire.
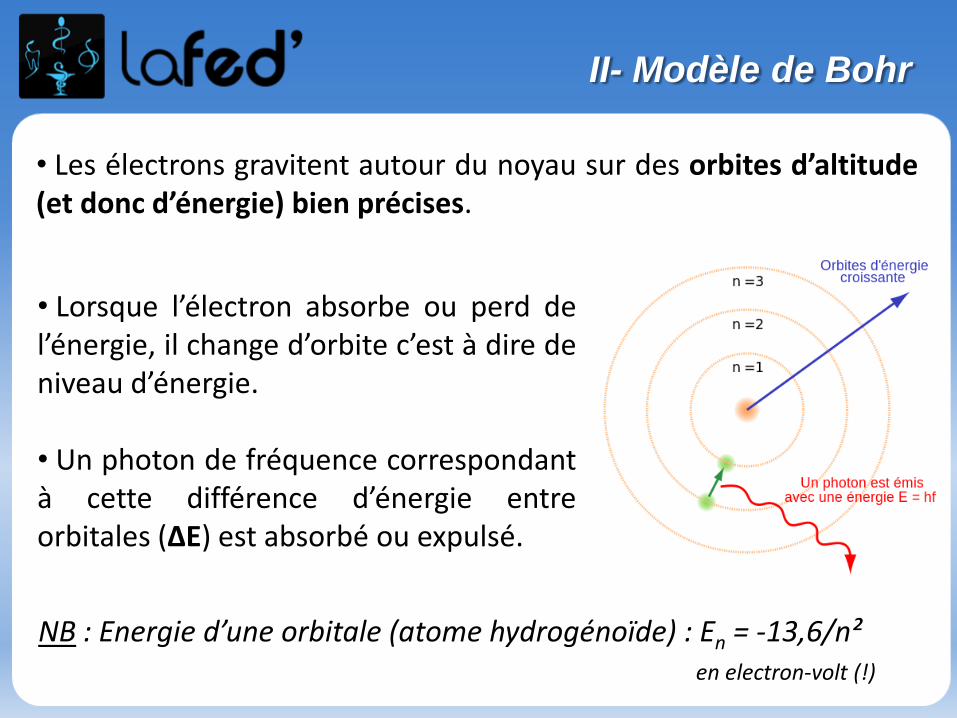
• Les électrons gravitent autour du noyau sur des orbites d’altitude(et donc d’énergie) bien précises.
• Lorsque l’électron absorbe ou perd del’énergie, il change d’orbite c’est à dire deniveau d’énergie.
• Un photon de fréquence correspondantà cette différence d’énergie entreorbitales (∆E) est absorbé ou expulsé.
NB : Energie d’une orbitale (atome hydrogénoïde) : En = -13,6/n²en electron-volt (!)
II- Modèle de Bohr
18
II- Modèle de Bohr

Exercices Votre premier QCM d’UE1 Répondre Vrai ou Faux pour chaque item :
A. La lumière peut s’échanger sous la forme de grains élémentaires appelés « photons » B. Le domaine du visible correspond au domaine du spectre électromagnétique compris
entre 400 et 750 nm. C. La relation de Planck s’écrit E = h.ν dans laquelle E est exprimée en électron-volt.D. Pour passer de la couche L à la couche K, un électron absorbe de l’énergie.E. Le modèle de Bohr est totalement « has been ».
Lorsqu’un atome d’hydrogène se désexcite, quel spectre lumineux émet-il ?
Celui-ci ? Ou bien celui-là ?
Un électron peut-il passer d’un niveau d’énergie En = -13,6 eV à un niveau En = - 3,4 eV si on l’expose à une source de photons d’énergie E = 20,0 eV ?
II- Modèle de Bohr

Plus complexe, il est la base de la compréhension de la chimie moderne générale comme organique : son étude est incontournable.
Un électron peut-être également assimilé à une onde. A l’échelle atomique, celarevient à abandonner définitivement l’idée de trajectoire et à faire un effortd’abstraction.
Ce qu’on peut définir = Probabilité de présence. Ce modèle est essentiellementmathématique.
III- Modèle quantique de l’atome
Limites du modèle de Bohr
Modèle quantique de l’atome
Conception « planétaire » de l’atome : séduisante mais à rejeter car ne permetpas de rendre compte de tous les faits expérimentaux, notamment magnétiques.

III- Modèle quantique de l’atome
Les 4 nombres quantiques
La probabilité de présence d’un électron est définie par sa fonction d’onde.
Cette fonction d’onde est reliée à son énergie par l’équation de Schrödinger. Larésolution de cette équation différentielle fait apparaître 3 nombres quantiques :
Ces trois nombres quantiques permettent de définir une orbitale atomique :volume dans lequel on a 95% de probabilité de retrouver l’électron à tout instant
Il existe un quatrième nombre quantique, relatif à une propriété intrinsèque de
l’électron et sans rapport avec l’orbitale décrite. s: nombre quantique de spin
n : nombre quantique principall : nombre quantique secondaire (ou azimutal)m : nombre quantique magnétique

Les électrons se répartissent autour du noyau dans des orbitales = structure électronique. Un électron est défini par 4 nombres quantiques (n, l, m et s).
Symboles Nom Valeurs permises
n nbre quantique principal : détermine
la couche (n=1 : couche K, n=2 = couche L, etc)
1, 2, 3, etc…
l nbre quantique secondaire : détermine la
sous-couche, forme de l’orbitale
de 0 à n-1
m nbre quantique magnétique : détermine
l’orientation de l’orbitale dans l’espace
- l ≤ m ≤ + l
s nbre quantique de spin : rotation de
l’électron sur lui-même, ↑ (+ ½) ou ↓ (- ½)
+ ½ ou - ½
III- Modèle quantique de l’atome
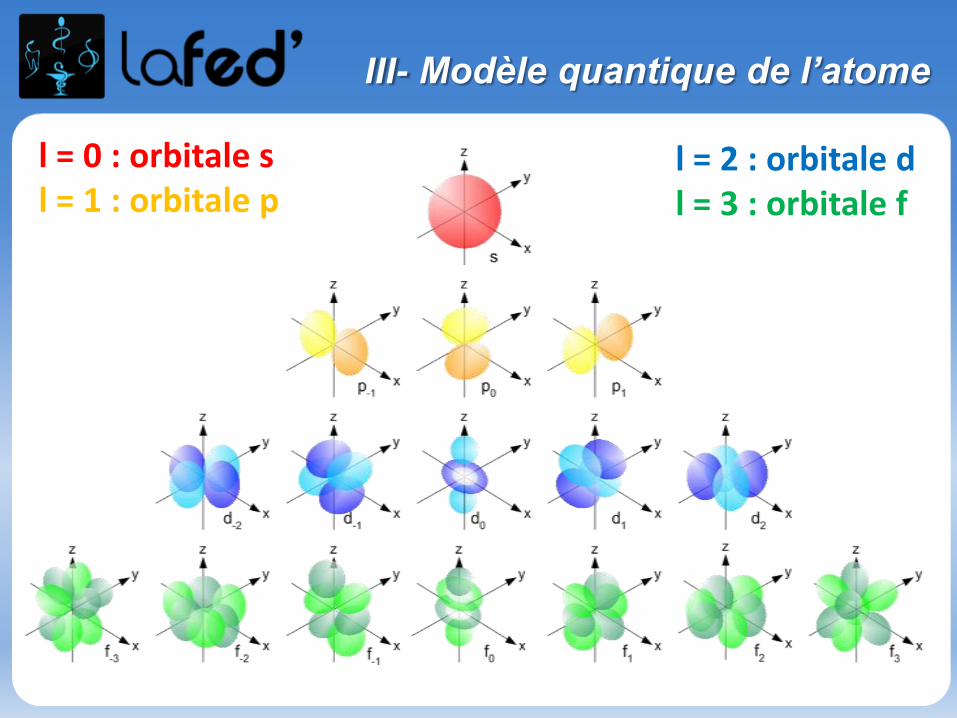
III- Modèle quantique de l’atome
l = 0 : orbitale sl = 1 : orbitale p
l = 2 : orbitale dl = 3 : orbitale f
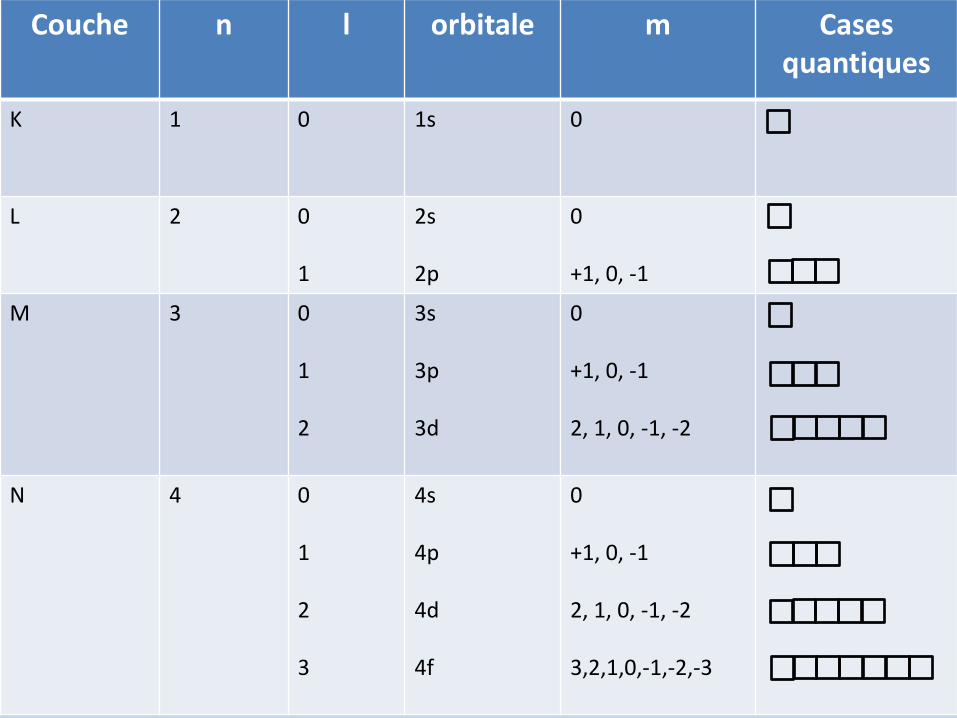
Couche n l orbitale m Casesquantiques
K 1 0 1s 0
L 2 0
1
2s
2p
0
+1, 0, -1
M 3 0
1
2
3s
3p
3d
0
+1, 0, -1
2, 1, 0, -1, -2
N 4 0
1
2
3
4s
4p
4d
4f
0
+1, 0, -1
2, 1, 0, -1, -2
3,2,1,0,-1,-2,-3

III- Modèle quantique de l’atome
Energie des orbitales atomiques
E (n, l) = - 13,6 x (Z-σ)²/n²
L’énergie d’un électron dépend de n et de l, par :
- L’attraction du noyau : fonction de la distance, nombre quantique n- La répulsion des autres électrons : via une constant d’écran σ, quivient diminuer la charge positive « Z » du noyau.
→ σ est fonction de n (altitude de l’OA) et de l (géométrie de l’OA)
Pour info :
Pour un atome hydrogénoïde : σ = 0, l’énergie ne dépend que de n→ On retrouve le modèle de Bohr.
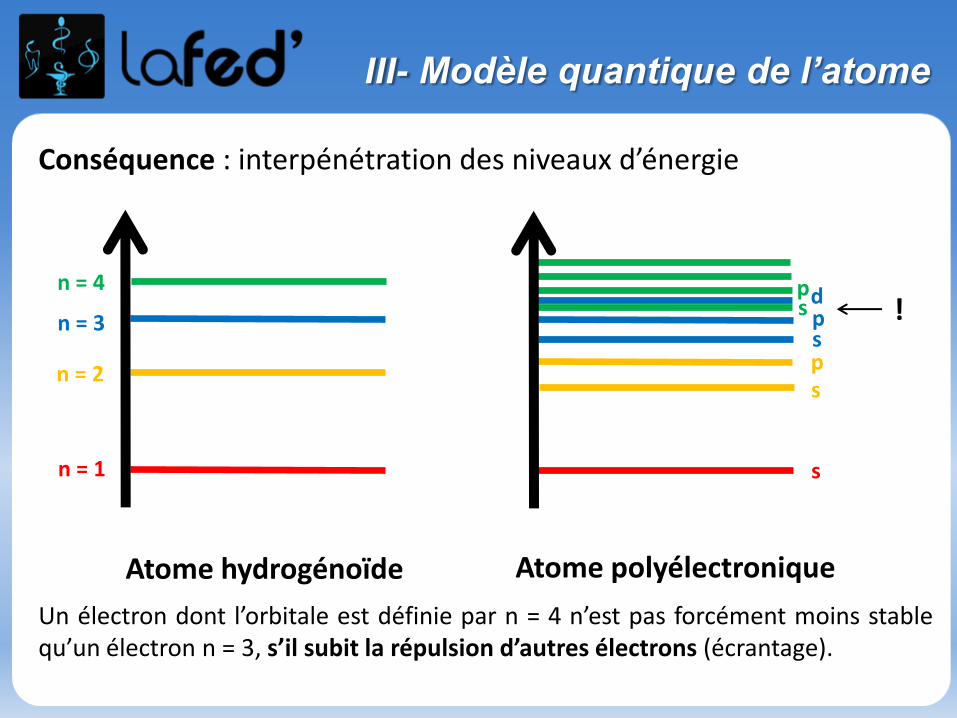
III- Modèle quantique de l’atome
Conséquence : interpénétration des niveaux d’énergie
Un électron dont l’orbitale est définie par n = 4 n’est pas forcément moins stablequ’un électron n = 3, s’il subit la répulsion d’autres électrons (écrantage).
n = 1
n = 2
n = 3
n = 4
s
spspds
Atome hydrogénoïde Atome polyélectronique
!p

Exercices Quel nombre quantique définit :
- L’orientation d’une orbitale dans l’espace ?- La couche dans laquelle est comprise l’orbitale ? - La forme d’une orbitale ?- Le spin d’un électron ?
Soit l’orbitale atomique suivante :
- Est-ce une orbitale s ou une orbitale p ?- Peut-elle être définie par le triplet de nombre quantique : n = 1 ; l = 1 ; m = 0 ?- Combien peut-elle prendre d’orientations dans l’espace ?- Au fait, c’est quoi une orbitale atomique ?
De quels nombres quantiques dépend l’énergie d’une orbitale :
- Pour un atome hydrogénoïde ?- Pour un atome polyélectronique ?
III- Modèle quantique de l’atome

IV- Configuration électronique
Configuration électronique de l’atome
Nous avons modélisé un certain nombres d’orbitales : il faut àprésent étudier comment on les remplit et dans quel ordre.
Rappel : une orbitale est défini par n, l, mun électron est défini par son orbitale (n, l, m) et son spin s
On représente une orbitale par une case quantique.
Elle peut être vide , demi-saturée , saturée (2 e-)
Remplissage des OA : 3 règles à connaître (Klechkowski, Hund, Pauli)
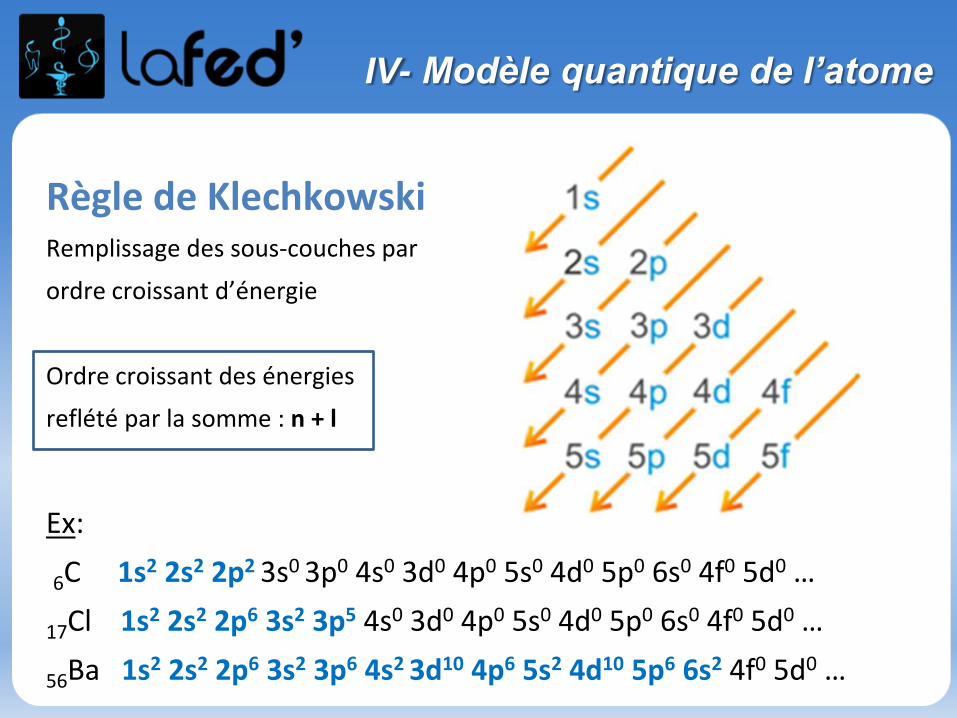
IV- Modèle quantique de l’atome
Règle de KlechkowskiRemplissage des sous-couches par
ordre croissant d’énergie
Ordre croissant des énergies
reflété par la somme : n + l
Ex:
6C 1s2 2s2 2p2 3s0 3p0 4s0 3d0 4p0 5s0 4d0 5p0 6s0 4f0 5d0 …
17Cl 1s2 2s2 2p6 3s2 3p5 4s0 3d0 4p0 5s0 4d0 5p0 6s0 4f0 5d0 …
56Ba 1s2 2s2 2p6 3s2 3p6 4s2 3d10 4p6 5s2 4d10 5p6 6s2 4f0 5d0 …
IV- Configuration électronique
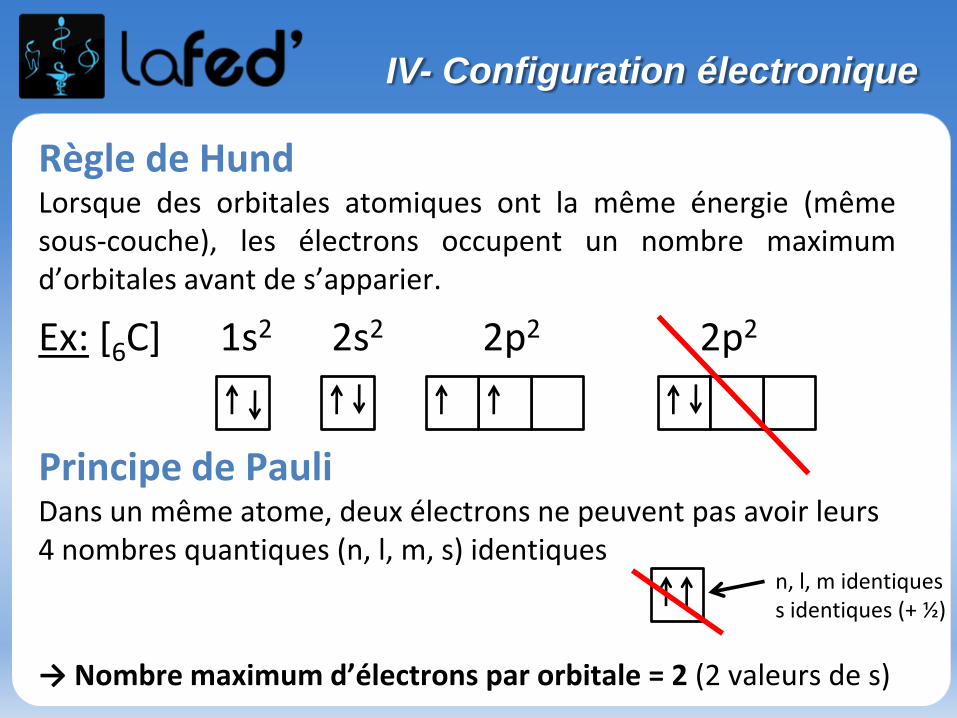
Règle de HundLorsque des orbitales atomiques ont la même énergie (mêmesous-couche), les électrons occupent un nombre maximumd’orbitales avant de s’apparier.
Ex: [6C] 1s2 2s2 2p2 2p2
Principe de PauliDans un même atome, deux électrons ne peuvent pas avoir leurs 4 nombres quantiques (n, l, m, s) identiques
→ Nombre maximum d’électrons par orbitale = 2 (2 valeurs de s)
n, l, m identiquess identiques (+ ½)
IV- Configuration électronique
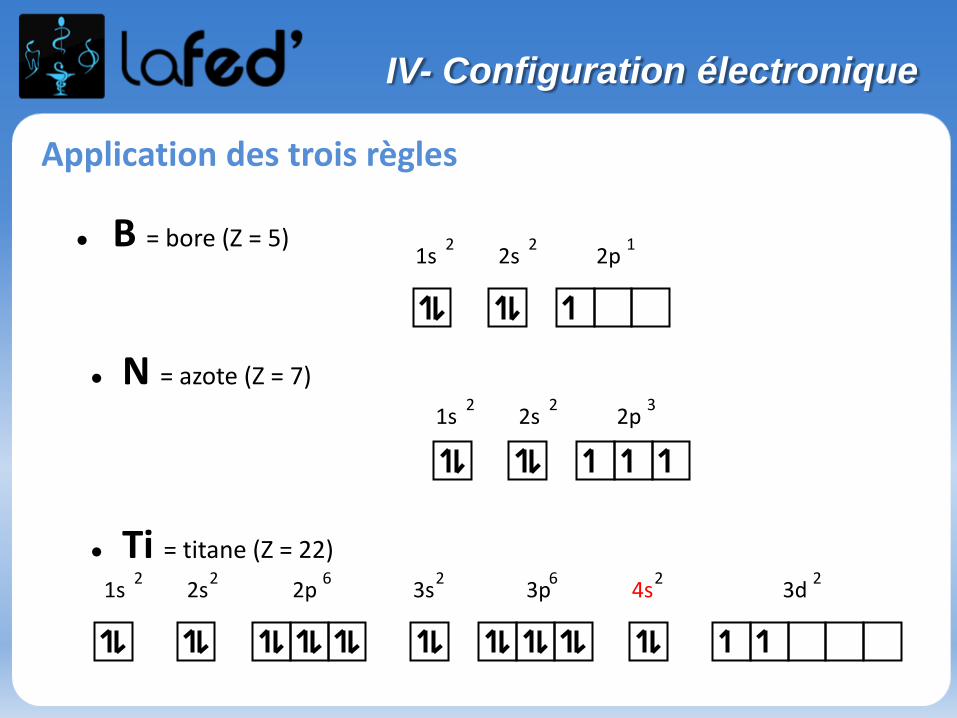
1s 2s 2p2 2 1
1s 2s 2p2 2 3
1s 2s 2p 3s 3p 4s 3d 2 2 6 2 6 2 2
● B = bore (Z = 5)
● N = azote (Z = 7)
● Ti = titane (Z = 22)
Application des trois règles

Cations et Anions (Atomes ayant perdu/gagné des électrons)
CATIONS : les électrons arrachés en premier sont les plus externes (cad sur le niveau n le plus grand)
Ex: [26Fe] = [Ar] 3d6 4s2
[26Fe2+] = [Ar] 3d6 4s0
[26Fe3+] = [Ar] 3d5 4s0
ANIONS : le remplissage se fait normalement
Ex: [17Cl] = [Ne] 3s2 3p5
[17Cl-] = [Ne] 3s2 3p6 (= [Ar] )
IV- Configuration électronique

Exercices
A. Une orbitale atomique peut accueillir un maximum de 2 électrons.B. Selon la règle de Klechkowski, on remplit les orbitales par ordre de « n+l » croissant.C. Selon la règle de Hund, deux électrons ne peuvent pas avoir leurs 4 nombres quantiques
identiques.D. La structure électronique du nickel (Z = 28) est 1s2 2s2 2p6 3s2 3p6 4s2 3d8
E. La structure électronique de l’ion Ni2+ est 1s2 2s2 2p6 3s2 3p6 4s2 3d6
Le jour J, on demande dans un QCM la configuration du carbone (Z=6) à l’état fondamental.Trois PACES (qui n’auront pas leur concours) choisissent ces configurations électroniques :
Pourquoi ont-ils faux ?
1)
2)
3)
QCM : Gueuler « Vraaaaaiiiii » ou « Fauuuuuux » pour chaque item.
1s 2s 2p
1s 2s 2p
1s 2s 2p
IV- Configuration électronique

V- Tableau périodique
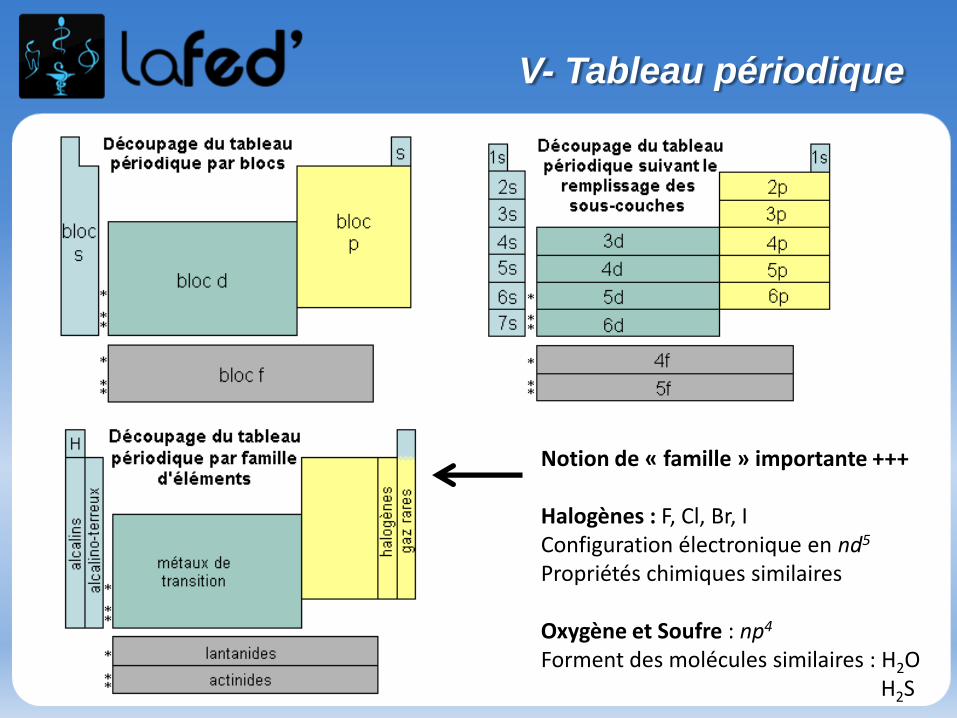
V- Tableau périodique
Notion de « famille » importante +++
Halogènes : F, Cl, Br, IConfiguration électronique en nd5
Propriétés chimiques similaires
Oxygène et Soufre : np4
Forment des molécules similaires : H2OH2S

Exercices
A quel bloc appartiennent : - Les halogènes ? - Les alcalins ?- Les lanthanides ?
Pouvez-vous citer les gaz rares dans l’ordre (sans regarder le tableau) ? Même question pour les halogènes.
A partir de quelle période voit-on apparaître les métaux de transition ?
Culture G : Savez-vous comment s’appelait le chimiste russe très intelligent qui a construit la première classification périodique ?
V- Tableau périodique

²Stage de Pré-Rentrée 2012UE 1 – Liaisons entre atomes
37
Diaporama réalisé par les tuteurs de La Fed’

I- Liaisons covalentes
Electrons de valenceOn appelle électrons de valence les e- qui vont pouvoir intervenir dans desliaisons = e- périphériques.
Définition
Une liaison covalente résulte de la mise en commun d’électrons de valenceentre deux (ou plusieurs) atomes ou ions.La distance inter-atomique idéale est celle pour laquelle l’édifice est le plus stable(il atteint le niveau de moindre énergie.)
L’état d’énergie entre l’état d’atomes séparés et l’état d’atomes liés représente àla fois l’énergie libérée lors de la liaison et l’énergie nécessaire pour rompre cetteliaison. C’est l’énergie de liaison (unité : J.mol-1 ou cal.mol-1)

A B. . A B..
A+ B-
A B..
Liaison covalente pure (cas limite)Résulte d’une forte interaction entre 2 atomes neutres où chacun fournit un e-. On a une paire d’e- partagée équitablement entre les 2 atomes.
Liaison covalente polaire (cas intermédiaire)Cas intermédiaire entre la covalente pure et l’ionique, où les atomes d’une molécule hétéronucléaire, entraînent un partage inégal des électrons.
Liaison ionique (cas limite)Formée par l’attraction de 2 atomes possédant unetrès grande différence d’électronégativité. Dans ce cas,il y a transfert d’un ou plusieurs e- , et formation d’ionsde charges opposées. (ex. Na+ et Cl-).
Les liaisons : caractère covalent et caractère électrostatique
I- Liaisons covalentes
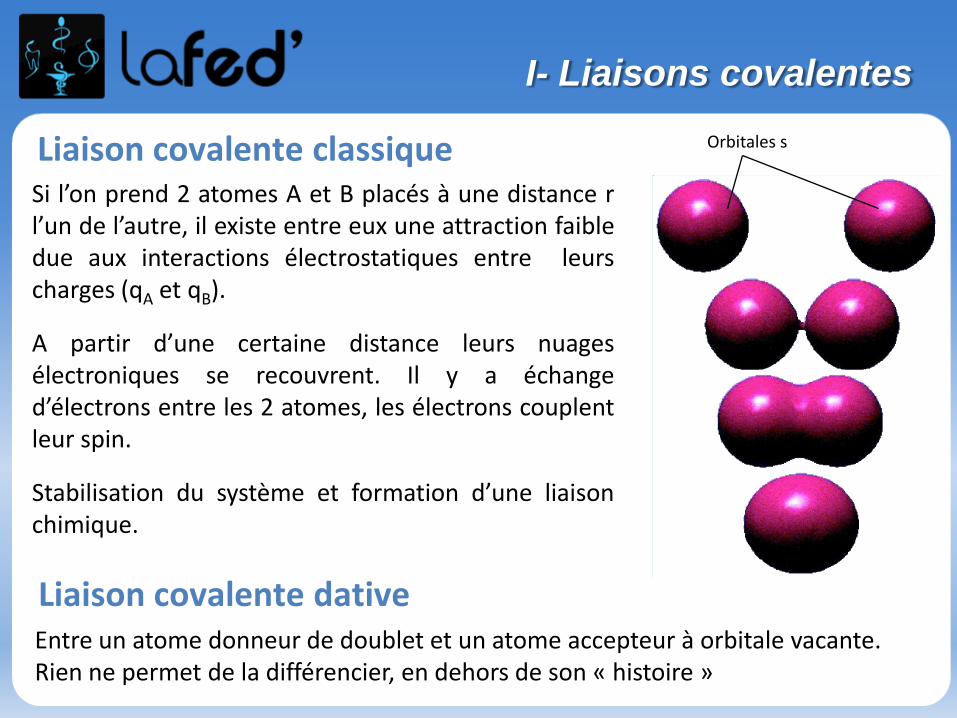
Orbitales s
Si l’on prend 2 atomes A et B placés à une distance rl’un de l’autre, il existe entre eux une attraction faibledue aux interactions électrostatiques entre leurscharges (qA et qB).
A partir d’une certaine distance leurs nuagesélectroniques se recouvrent. Il y a échanged’électrons entre les 2 atomes, les électrons couplentleur spin.
Stabilisation du système et formation d’une liaisonchimique.
Liaison covalente classique
Entre un atome donneur de doublet et un atome accepteur à orbitale vacante.Rien ne permet de la différencier, en dehors de son « histoire »
Liaison covalente dative
I- Liaisons covalentes
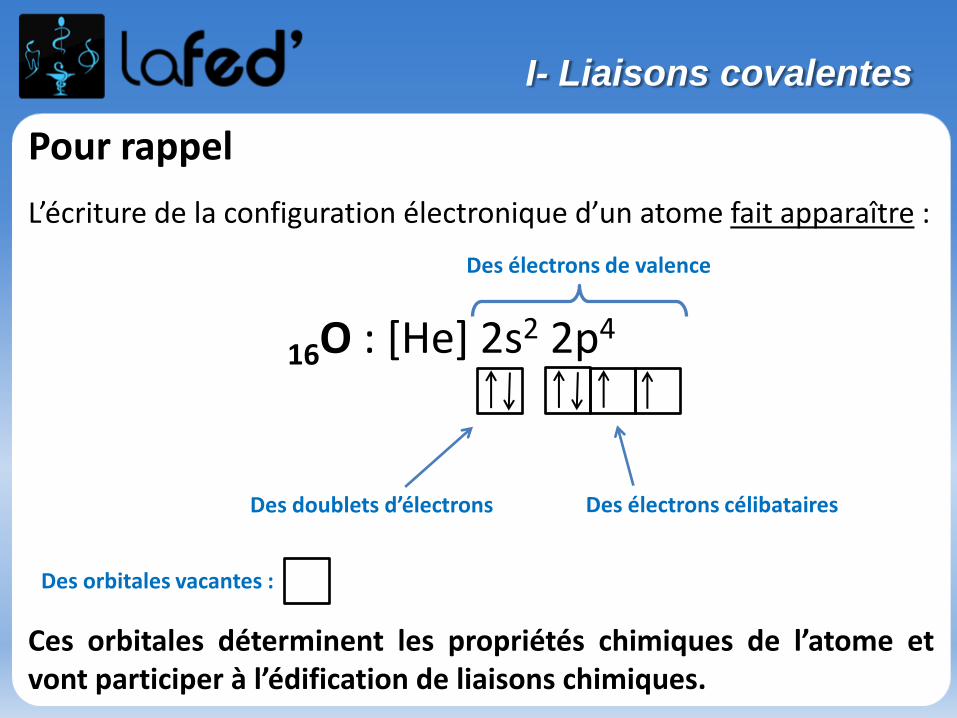
Pour rappel
L’écriture de la configuration électronique d’un atome fait apparaître :
16O : [He] 2s2 2p4
Ces orbitales déterminent les propriétés chimiques de l’atome etvont participer à l’édification de liaisons chimiques.
Des électrons de valence
Des doublets d’électrons Des électrons célibataires
Des orbitales vacantes :
I- Liaisons covalentes
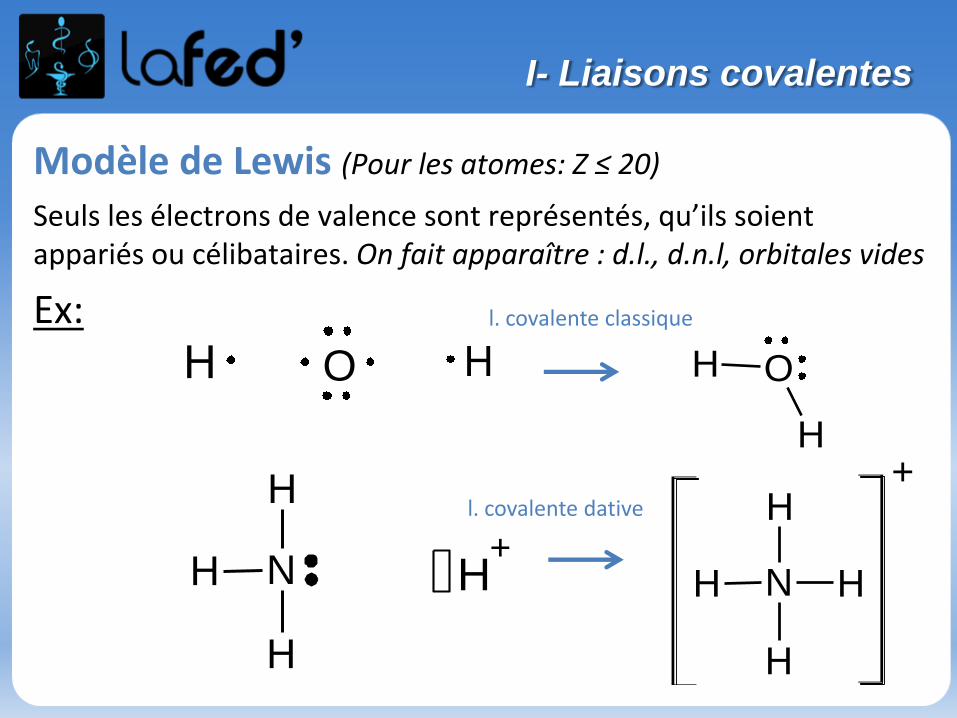
Modèle de Lewis (Pour les atomes: Z ≤ 20)
Seuls les électrons de valence sont représentés, qu’ils soient appariés ou célibataires. On fait apparaître : d.l., d.n.l, orbitales vides
Ex:
OH
H
O HH
N
H
H
H H+
N
H
H
H H
+
l. covalente classique
l. covalente dative
I- Liaisons covalentes

Règle de l’octet(Pour ns2 et npx dont Z ≤ 18)
Les atomes tendent à se combiner de façon à ce que leur coucheexterne renferme 8 e-. Un atome engagé dans une ou plusieursliaison(s) cherche à acquérir la configuration électronique dugaz rare qui le suit dans la classification périodique.
Ex:
I- Liaisons covalentes
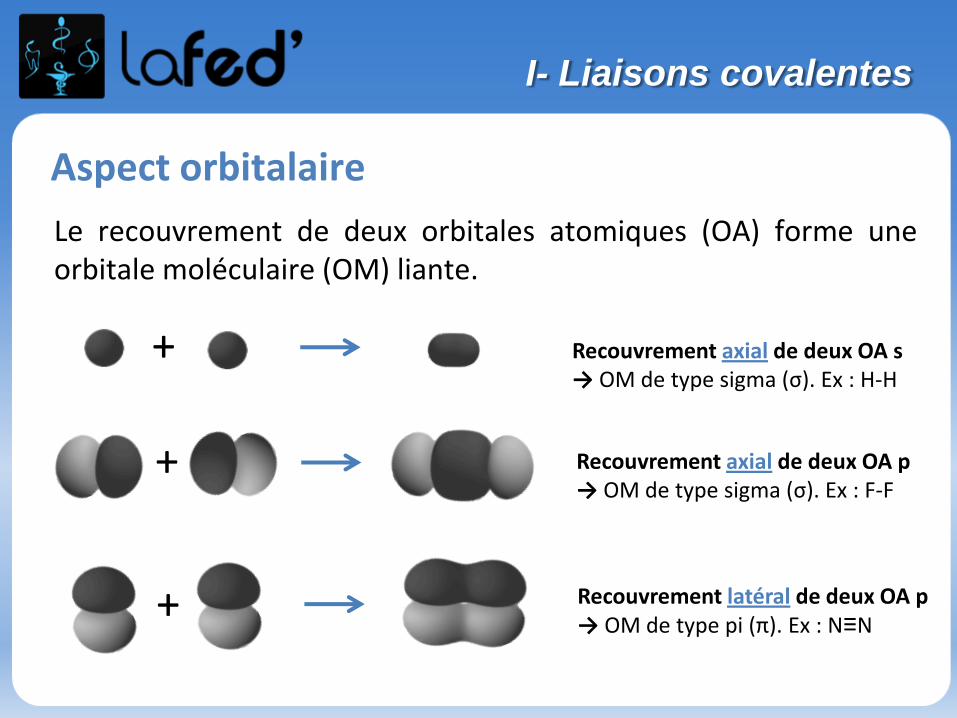
Aspect orbitalaire
Le recouvrement de deux orbitales atomiques (OA) forme uneorbitale moléculaire (OM) liante.
+
+
+
Recouvrement axial de deux OA s→ OM de type sigma (σ). Ex : H-H
Recouvrement axial de deux OA p→ OM de type sigma (σ). Ex : F-F
Recouvrement latéral de deux OA p→ OM de type pi (π). Ex : N≡N
I- Liaisons covalentes

Les liaisons peuvent être simples :
Les liaison peuvent être doubles ou triples :
1 recouvrement axial= Une liaison σ
CH3 CH3
CH2 CH2
CH CH
1 recouvrement axial (Un seul possible !)
+ un recouvrement latéral (= Une liaison σ + Une liaison π)
+ deux recouvrements latéraux(= Une liaison σ + Deux liaisons π)
I- Liaisons covalentes

Une liaison chimique naturelle, ça coûte de l’énergie ou bien ça dégage de la chaleur ?
La configuration électronique de l’azote s’écrit : *He+ 2s2 2p3
Combien de liaisons peut-il faire avec d’autres atomes ?Combien de doublets non liants ?
Soit deux atomes représentés par les points A et B. Les orbitales représentées de manière stylisée forment-t-elles des liaisons σ ou π ?Comment le reconnaître simplement ?
Exercices
I- Liaisons covalentes
A ● ● B A ● ● B

II- Le modèle de Gillespie
Prévision de la structure d’une molécule
Deux méthodes
1) Méthode de Gillespie ou « VSEPR »(VSEPR : Valence Shell Electron Pair Repulsion)
2) Méthode de l’hybridation
On doit arriver aux mêmes conclusions !

II- Le modèle de Gillespie
Règles de GillespieUne molécule peut être notée de la manière suivante:
A Xm En
Atome central
Atomes liés, m = nombre(peuvent être de nature ≠)
Doublets non liantsn = nombre

Les m doublets liants (DL) et n doublets non-liants (DnL) secomportent comme des charges négatives.
Les doublets d’e- se placent de manière à ce que lesrépulsions électroniques soient minimales.
Lorsqu’un atome X est lié à l’atome central par une double ouune triple liaison, la géométrie est pratiquement la même qu’enprésence d’une simple liaison entre les deux atomes.
II- Le modèle de Gillespie
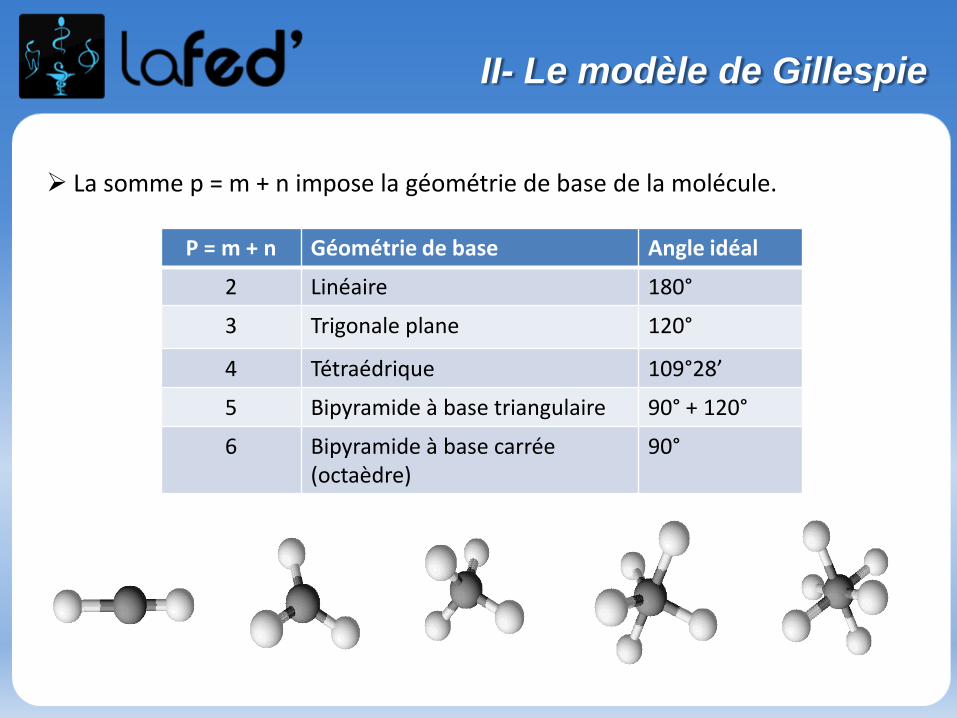
La somme p = m + n impose la géométrie de base de la molécule.
P = m + n Géométrie de base Angle idéal
2 Linéaire 180°
3 Trigonale plane 120°
4 Tétraédrique 109°28’
5 Bipyramide à base triangulaire 90° + 120°
6 Bipyramide à base carrée (octaèdre)
90°
II- Le modèle de Gillespie

Exemple 1 : Déterminer la géométrie du soufre dans SO2
16S : [Ne] 3s2 3p4
→ 6 électrons de valence→ 4 électrons utilisés pour liaison avec O→ Reste 2 électrons = 1 d.n.l. → Conclusion : m+n = 3; trigonal plan (AX2E)
Exemple 2 : Déterminer la géométrie du phosphore dans PF5
15P : [Ne] 3s2 3p3
→ 5 électrons de valence→ 5 électrons utilisés pour liaison avec F→ Reste 0 électrons = 0 d.n.l. → Conclusion : m+n = 5; bipyramide à base ∆ (AX5)
SO O
P
F
F
FF
F
II- Le modèle de Gillespie
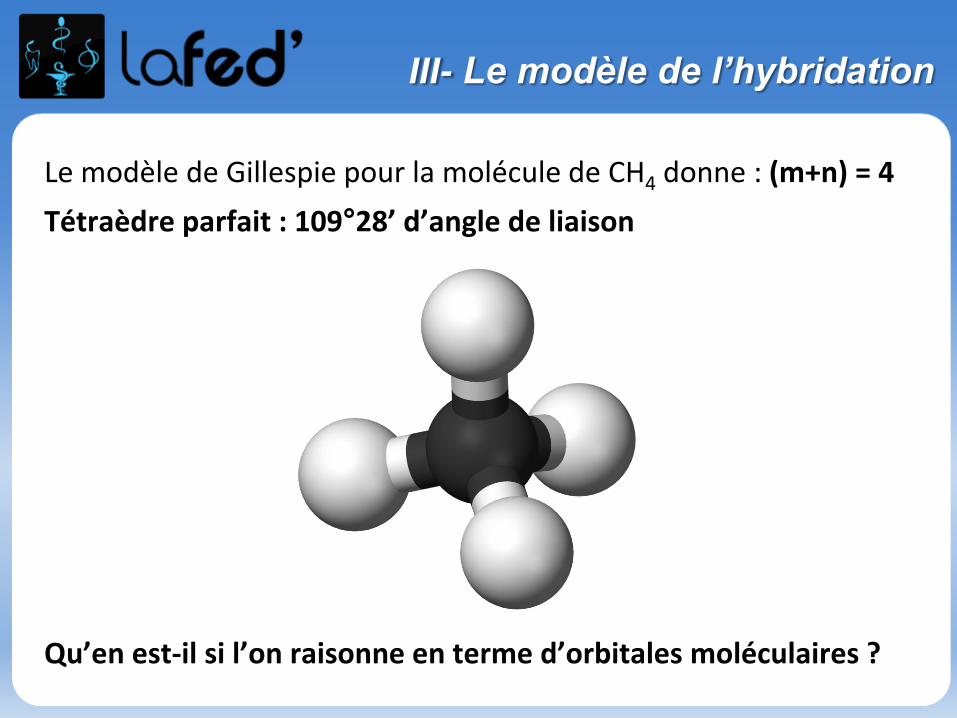
III- Le modèle de l’hybridation
Le modèle de Gillespie pour la molécule de CH4 donne : (m+n) = 4
Tétraèdre parfait : 109°28’ d’angle de liaison
Qu’en est-il si l’on raisonne en terme d’orbitales moléculaires ?
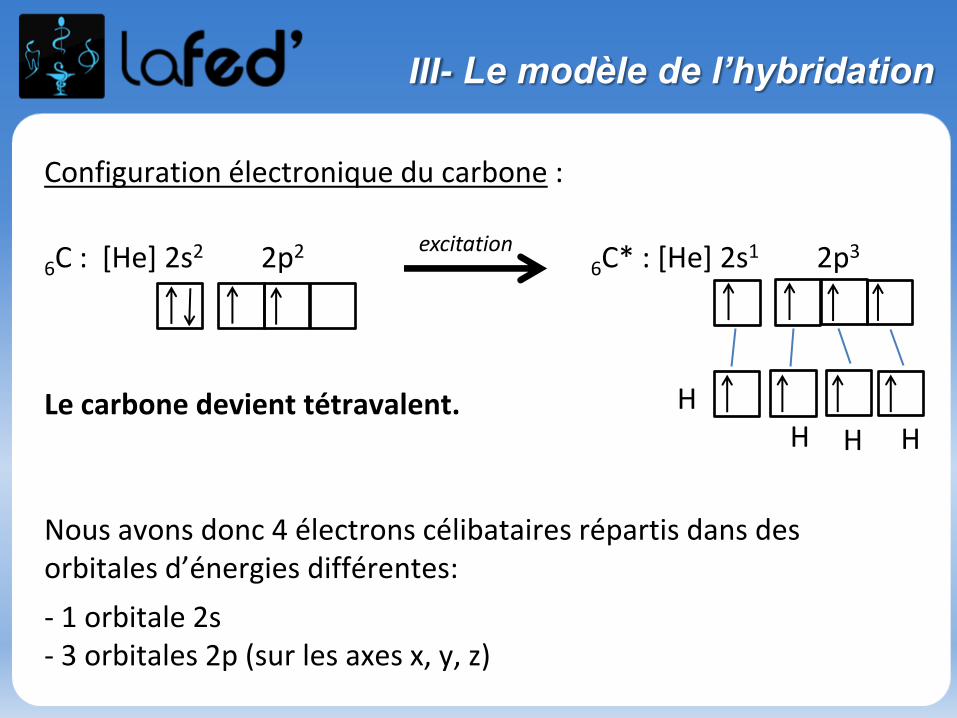
III- Le modèle de l’hybridation
Configuration électronique du carbone :
6C : [He] 2s2 2p26C* : [He] 2s1 2p3
Le carbone devient tétravalent.
Nous avons donc 4 électrons célibataires répartis dans des orbitales d’énergies différentes:
- 1 orbitale 2s- 3 orbitales 2p (sur les axes x, y, z)
excitation
H
HHH
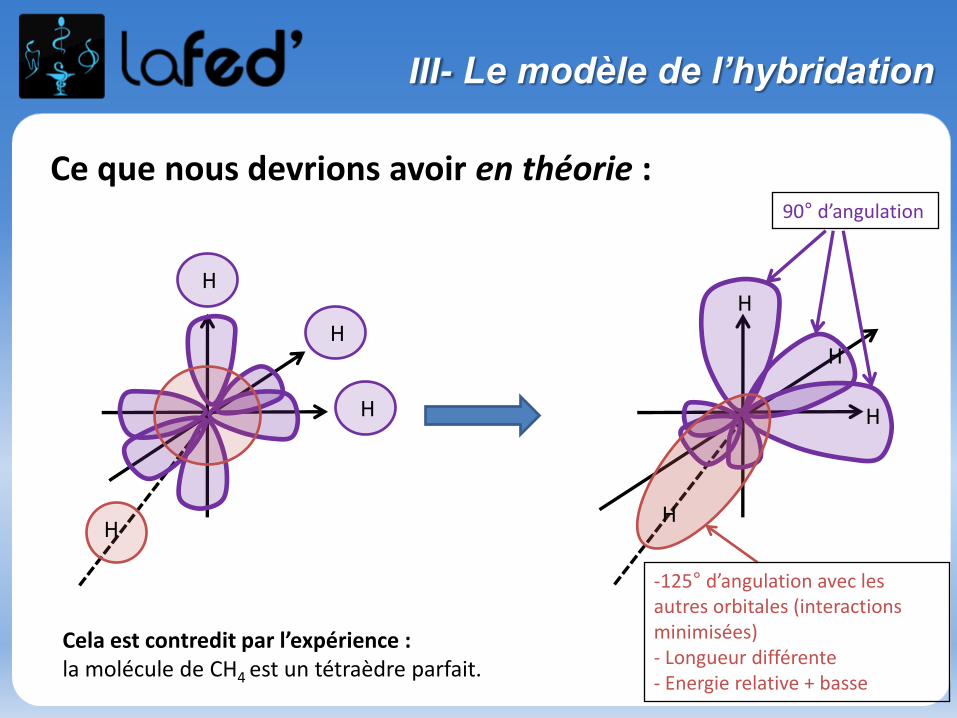
-125° d’angulation avec les autres orbitales (interactions minimisées)- Longueur différente- Energie relative + basse
90° d’angulation
H
H
H
HH
H
H
H
Ce que nous devrions avoir en théorie :
Cela est contredit par l’expérience : la molécule de CH4 est un tétraèdre parfait.
III- Le modèle de l’hybridation
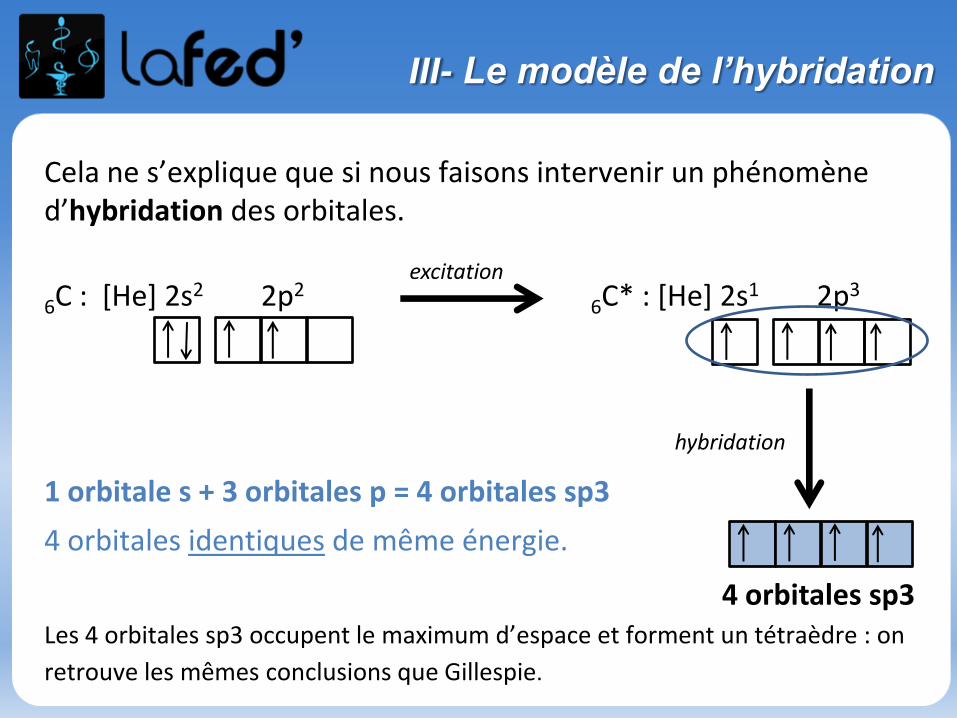
III- Le modèle de l’hybridation
Cela ne s’explique que si nous faisons intervenir un phénomène d’hybridation des orbitales.
6C : [He] 2s2 2p26C* : [He] 2s1 2p3
1 orbitale s + 3 orbitales p = 4 orbitales sp3
4 orbitales identiques de même énergie.
Les 4 orbitales sp3 occupent le maximum d’espace et forment un tétraèdre : on
retrouve les mêmes conclusions que Gillespie.
excitation
hybridation
4 orbitales sp3

hybridation Géométrie de base Angle idéal
sp Linéaire 180°
sp2 Trigonale plane 120°
sp3 Tétraédrique 109°28’
sp3d Bipyramide à base triangulaire 90° + 120°
sp3d2 Bipyramide à base carrée (octaèdre)
90°
Différents type d’hybridation :
Attention !
On observe l’hybridation que pour les liaisons σ.
JAMAIS pour les liaisons π !
III- Le modèle de l’hybridation
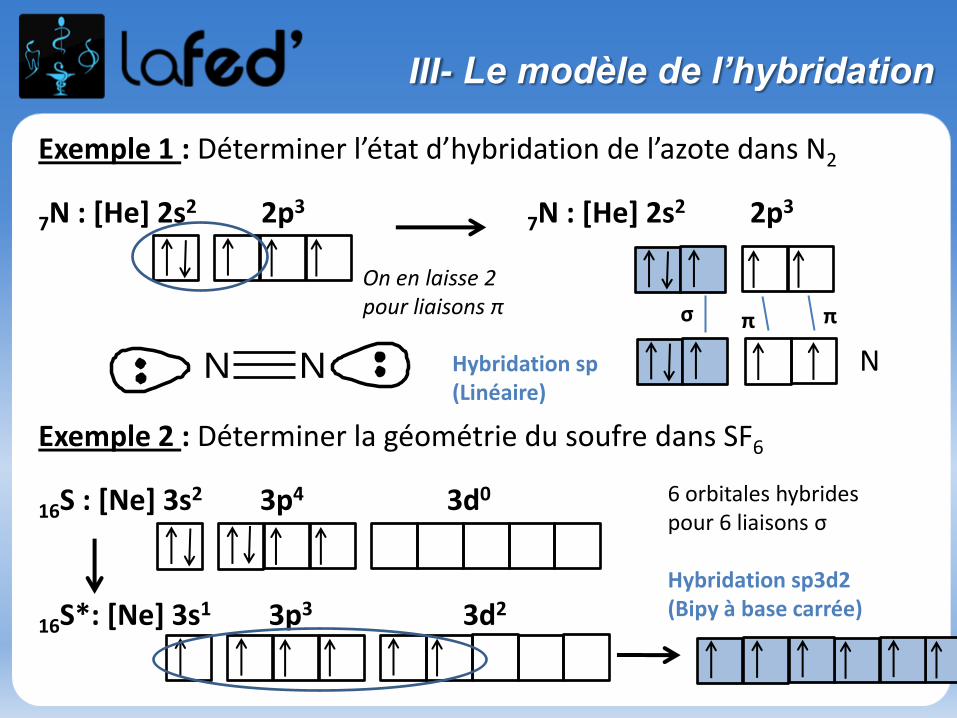
Exemple 1 : Déterminer l’état d’hybridation de l’azote dans N2
7N : [He] 2s2 2p37N : [He] 2s2 2p3
Exemple 2 : Déterminer la géométrie du soufre dans SF6
16S : [Ne] 3s2 3p4 3d0
16S*: [Ne] 3s1 3p3 3d2
σ π π
Hybridation sp(Linéaire)
N N
On en laisse 2 pour liaisons π
N
6 orbitales hybrides pour 6 liaisons σ
Hybridation sp3d2 (Bipy à base carrée)
III- Le modèle de l’hybridation
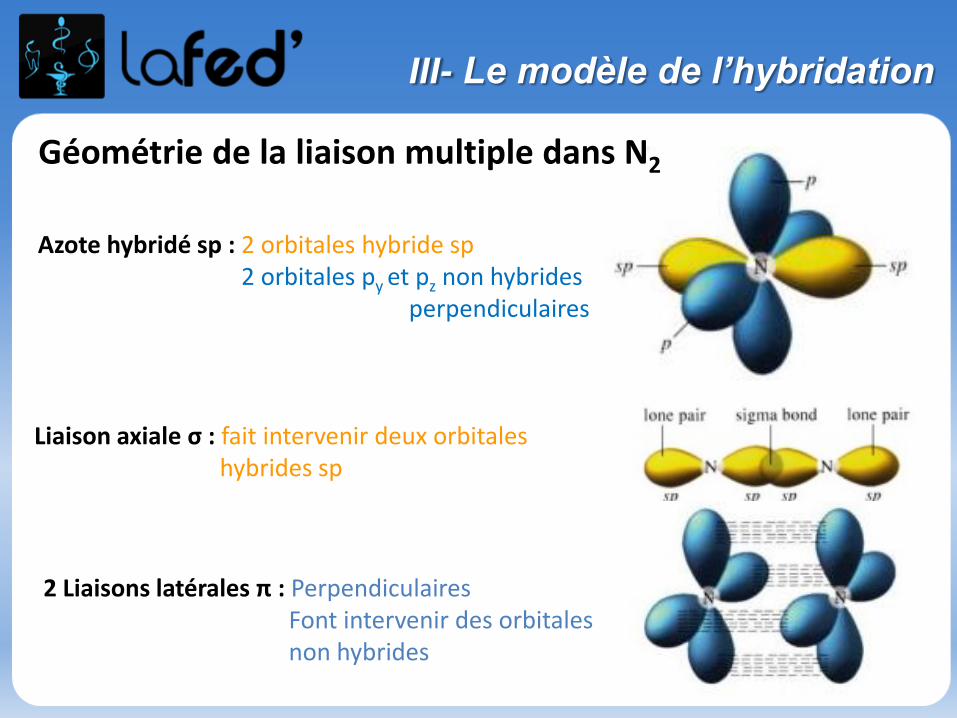
Géométrie de la liaison multiple dans N2
Azote hybridé sp : 2 orbitales hybride sp2 orbitales py et pz non hybrides
perpendiculaires
Liaison axiale σ : fait intervenir deux orbitales hybrides sp
2 Liaisons latérales π : PerpendiculairesFont intervenir des orbitales non hybrides
III- Le modèle de l’hybridation

QCM : Nous vous prions de bien vouloir avoir l’amabilité d’exprimer votre avis au plus vite concernant les quelques propositions ci-dessous. Bien cordialement, vos Tuteurs d’UE1.
A. Le modèle de Gillespie est fondé sur la répulsion des paires électroniques.B. Une molécule AX3E dans le modèle de Gillespie prendra une forme trigonale plane.C. Dans le modèle de l’hybridation : 3 orbitales « p » + 1 orbitale « s » = 1 orbitale « sp3 ».D. Un atome hybridé sp prendra une forme linéaire.E. Les orbitales moléculaires π sont toujours exclues de l’hybridation.
Quel est l’angle idéal entre les liaisons d’un tétraèdre ?
Quel est l’angle entre deux orbitales moléculaires π dans la molécule de N2 ?Et entre deux orbitales moléculaires σ ?
Exercices
II- et III- Gillespie et Hybridation
IV- Application aux
molécules organiques
Molécules organiques :
Très répandues dans le vivant.Riches en C, H, O et N.
Vous avez maintenant les bases pour deviner la structure3D de presque toutes les molécules organiques.
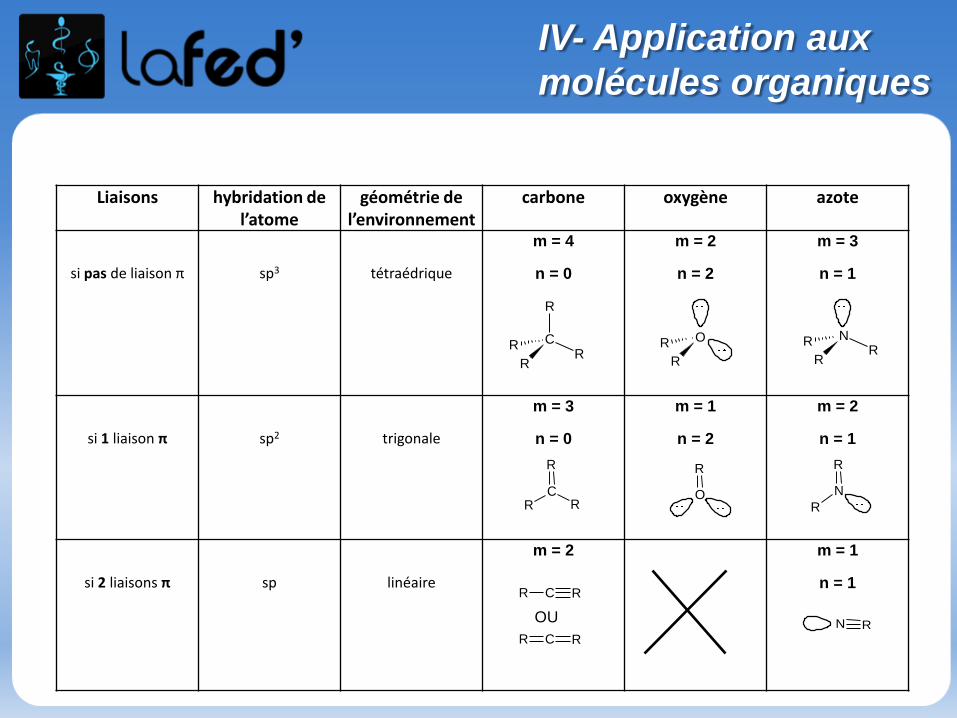
IV- Application aux
molécules organiques
Liaisons hybridation de l’atome
géométrie de l’environnement
carbone oxygène azote
si pas de liaison π sp3 tétraédrique
m = 4
n = 0
m = 2
n = 2
m = 3
n = 1
si 1 liaison π sp2 trigonale
m = 3
n = 0
m = 1
n = 2
m = 2
n = 1
si 2 liaisons π sp linéaire
m = 2 m = 1
n = 1
C
R
RR
RO
R
RN
RR
R
R
CRR
R
O
R
N
R
RCR
RCR
OURN
O
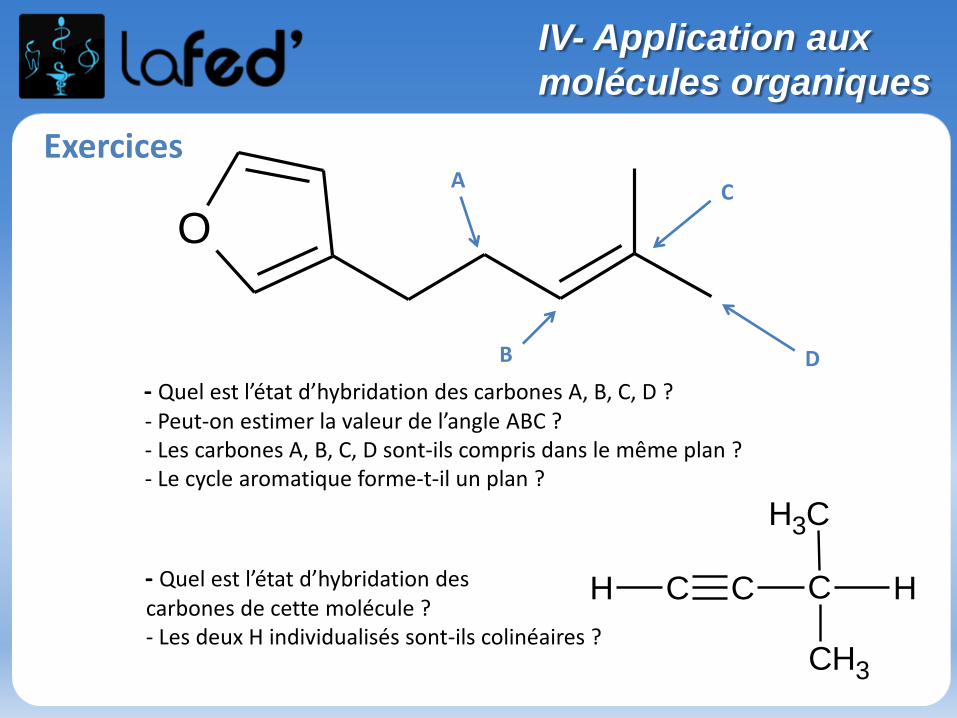
- Quel est l’état d’hybridation des carbones A, B, C, D ?- Peut-on estimer la valeur de l’angle ABC ?- Les carbones A, B, C, D sont-ils compris dans le même plan ?- Le cycle aromatique forme-t-il un plan ?
A
DB
C
- Quel est l’état d’hybridation des carbones de cette molécule ?- Les deux H individualisés sont-ils colinéaires ?
C C C
CH3
CH3
H H
IV- Application aux
molécules organiques
Exercices

Définition : liaisons faibles
Ce sont des liaisons non covalentes qui ne mettent donc en jeu ni échanges d’électrons ni recouvrements d’OA.
L’énergie mise en jeu est plus faible et la distance entre atomes est plus grande que pour des liaisons covalentes.
Quelques exemples d’interactions
Electrostatiques (Interactions entre charges ou dipôles)
Effet hydrophobe
V- Les liaisons intermoléculaires

Interactions électrostatiquesElles permettent la cohésion des liquides et des solides.
↗ des interactions ↗ T° de fusion
↗ T° d’ébullition
1) Interaction dipôle-dipôle
V- Les liaisons intermoléculaires
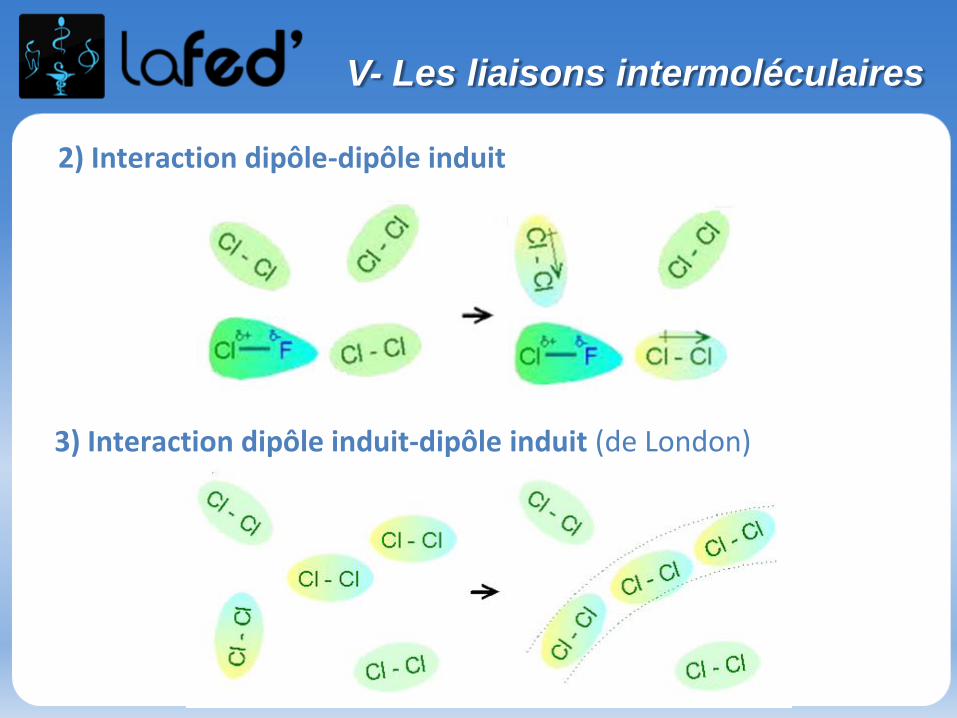
2) Interaction dipôle-dipôle induit
3) Interaction dipôle induit-dipôle induit (de London)
V- Les liaisons intermoléculaires
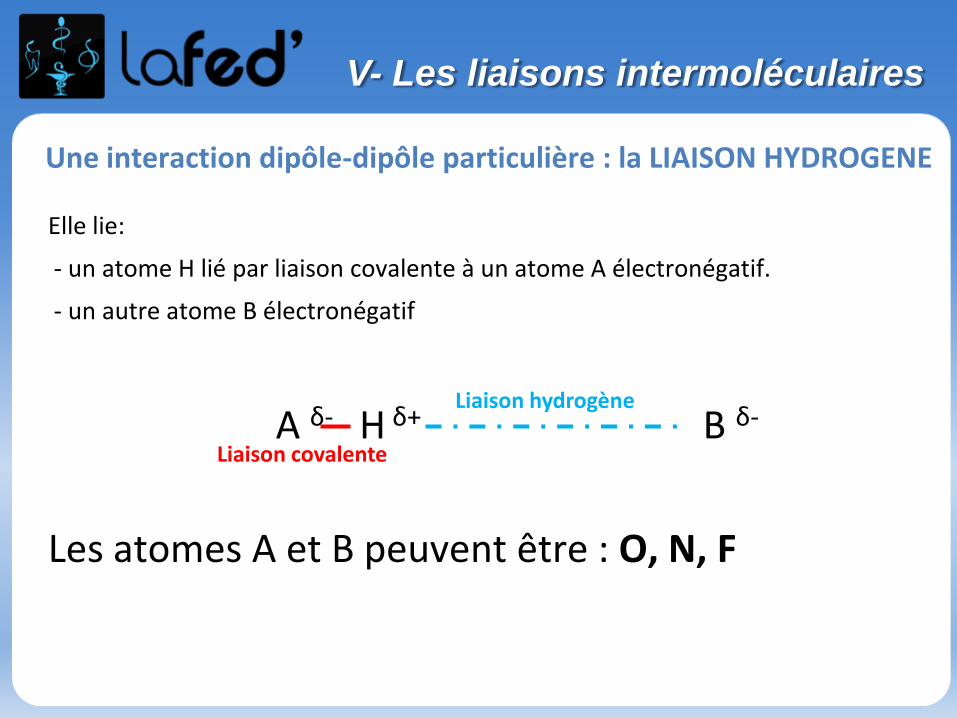
Une interaction dipôle-dipôle particulière : la LIAISON HYDROGENE
Elle lie:
- un atome H lié par liaison covalente à un atome A électronégatif.
- un autre atome B électronégatif
A δ- H δ+ B δ-
Les atomes A et B peuvent être : O, N, F
Liaison covalente
Liaison hydrogène
V- Les liaisons intermoléculaires
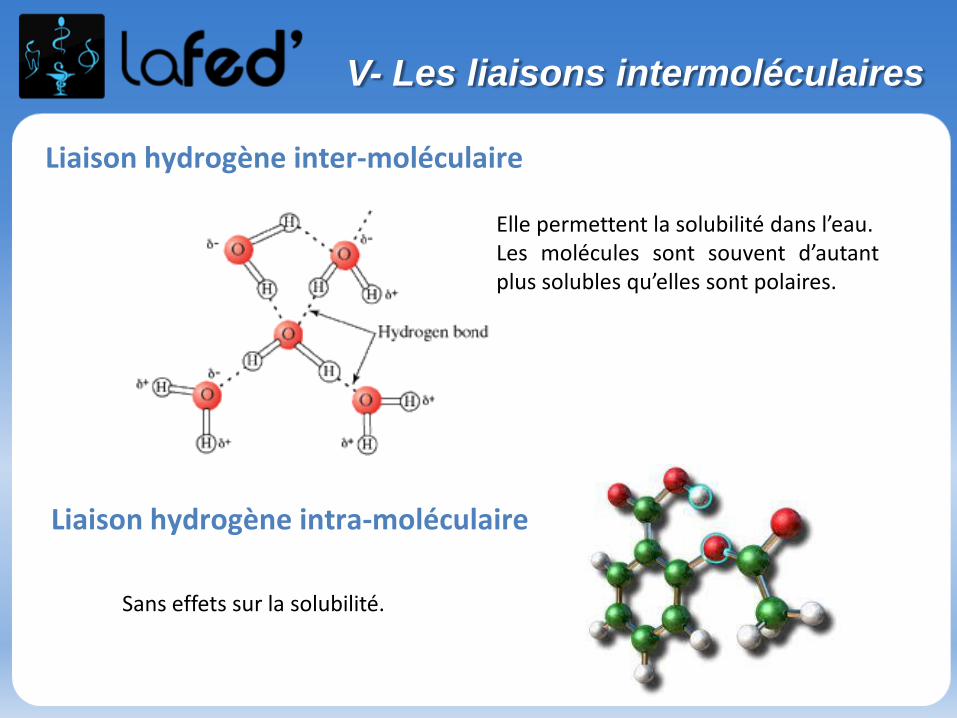
Liaison hydrogène inter-moléculaire
Elle permettent la solubilité dans l’eau.Les molécules sont souvent d’autantplus solubles qu’elles sont polaires.
Liaison hydrogène intra-moléculaire
Sans effets sur la solubilité.
V- Les liaisons intermoléculaires
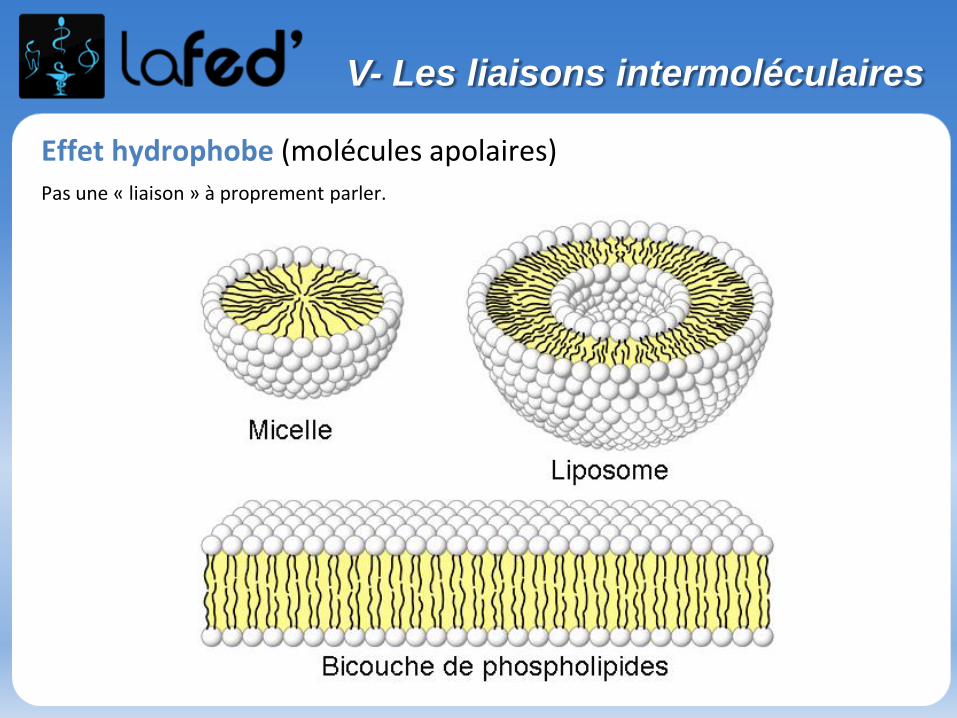
Effet hydrophobe (molécules apolaires)Pas une « liaison » à proprement parler.
V- Les liaisons intermoléculaires
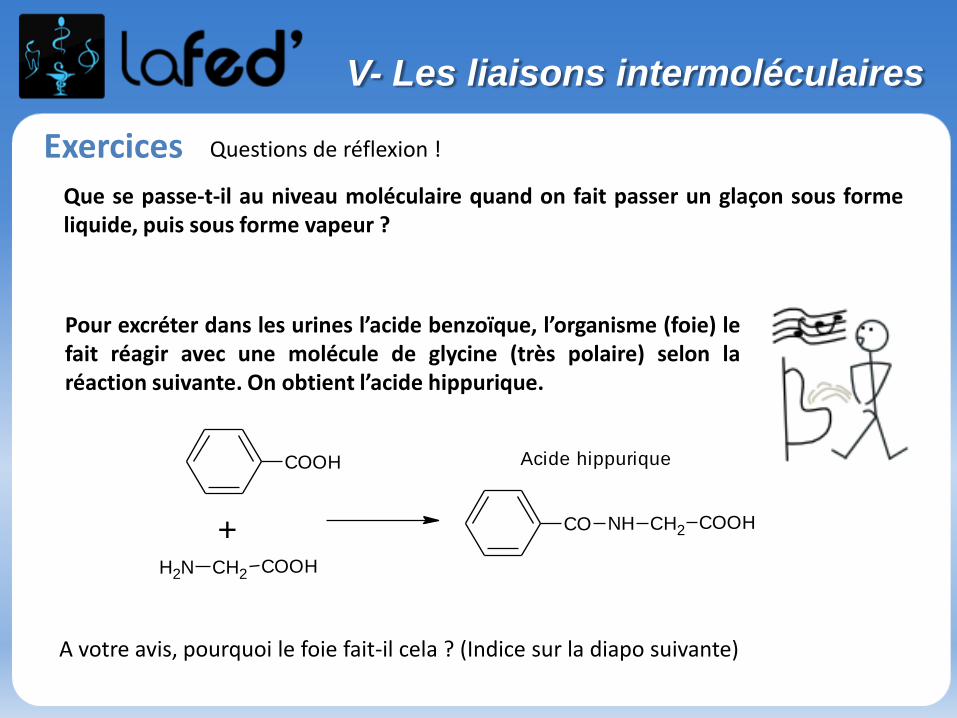
Exercices
V- Les liaisons intermoléculaires
Pour excréter dans les urines l’acide benzoïque, l’organisme (foie) lefait réagir avec une molécule de glycine (très polaire) selon laréaction suivante. On obtient l’acide hippurique.
+NH2 COOHCH2
COOH
CO NH COOHCH2
Acide hippurique
A votre avis, pourquoi le foie fait-il cela ? (Indice sur la diapo suivante)
Que se passe-t-il au niveau moléculaire quand on fait passer un glaçon sous formeliquide, puis sous forme vapeur ?
Questions de réflexion !
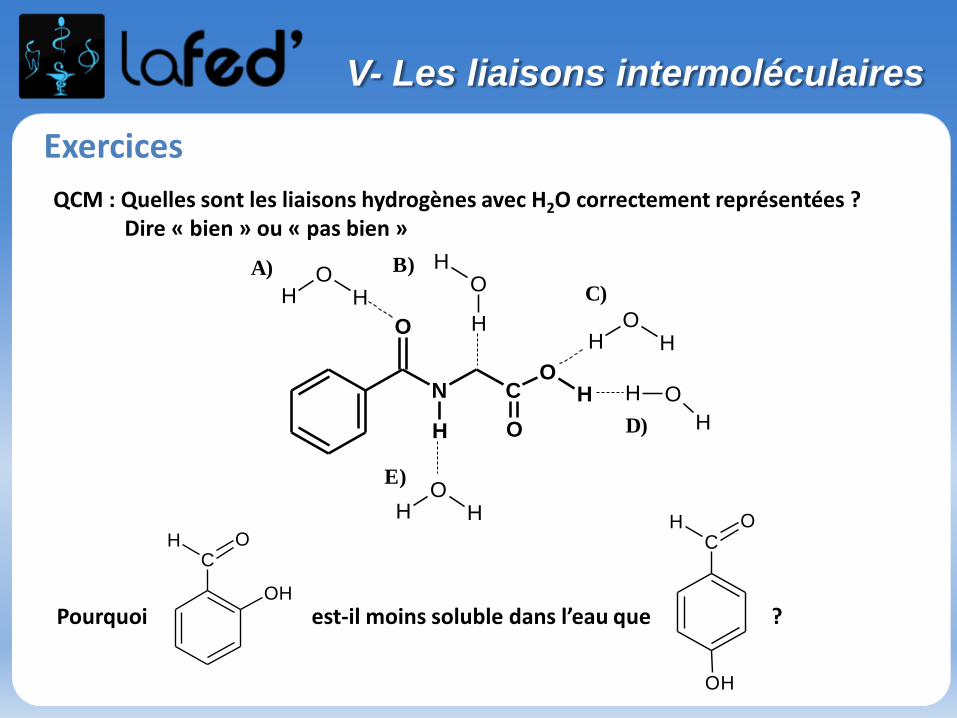
Exercices
V- Les liaisons intermoléculaires
QCM : Quelles sont les liaisons hydrogènes avec H2O correctement représentées ?Dire « bien » ou « pas bien »
N C
O
O
O
H
H
OHH
O
H
H
OHH
OHH
O
H
HA) B)
E)
C)
D)
Pourquoi est-il moins soluble dans l’eau que ?
C
OH
OH COH
OH

Stage de Pré-Rentrée 2012UE 1 – Isomérie et stéréochimie
71
Diaporama réalisé par les tuteurs de La Fed’

Formule brute :
- Peu informative- Indique seulement la composition atomique de la molécule
Exemples :
butane = C4H10
pentane = C5H12…
acide acétique = C2H4O2
éthanol = C2H6O
NB : molécules comprenant uniquement C et H = hydrocarbures
I- Représenter les molécules

Formule semi-developpée :
- L’organisation des atomes les uns par rapport aux autres(« qui est lié à qui ») en découle.- Mais la formule semi-développée, ne montre rien del’agencement des atomes dans l’espace tridimensionnel.
CH3 CH2 CH2 CH3 CH3 CH CH2 CH3
CH3
Exemples :
I- Représenter les molécules
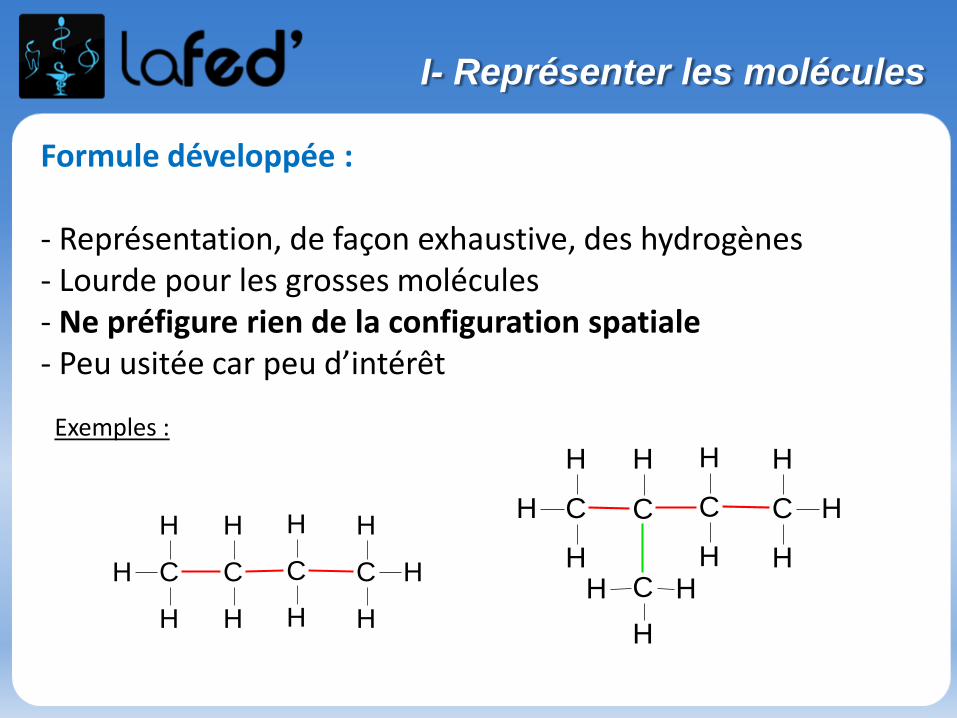
Formule développée :
- Représentation, de façon exhaustive, des hydrogènes- Lourde pour les grosses molécules- Ne préfigure rien de la configuration spatiale- Peu usitée car peu d’intérêt
C C C C
H
H
HH
HH
H
H
H
H
C C C C
H
H
HH
HH
C
H
H
H
H
H
H
Exemples :
I- Représenter les molécules
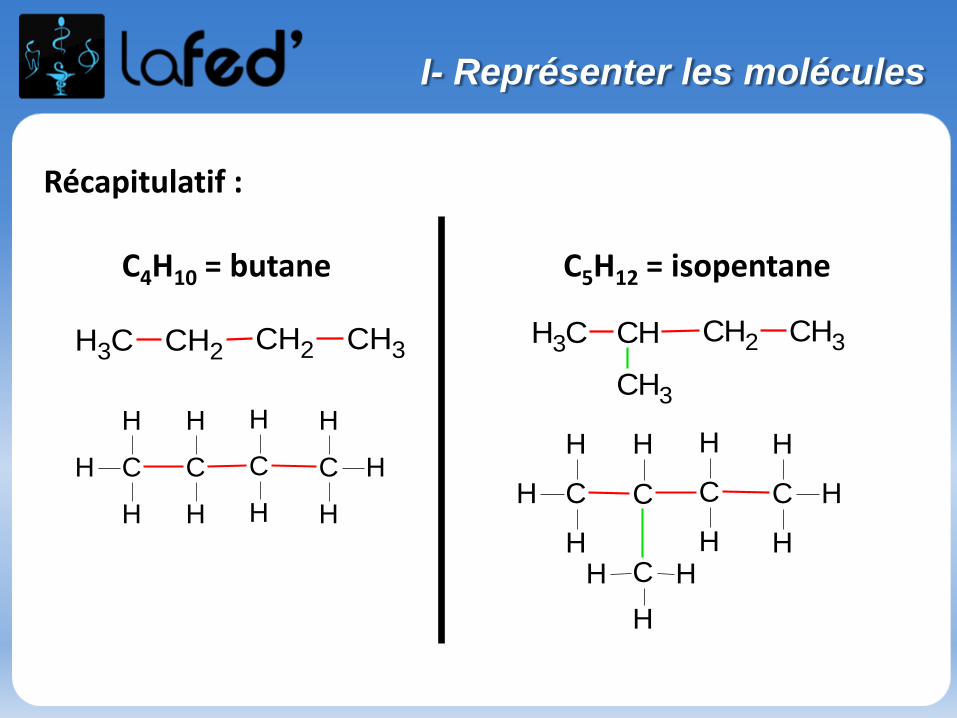
Récapitulatif :
C C C C
H
H
HH
HH
H
H
H
HC C C C
H
H
HH
HH
C
H
H
H
H
H
H
CH3 CH2 CH2 CH3CH3 CH CH2 CH3
CH3
C4H10 = butane C5H12 = isopentane
I- Représenter les molécules

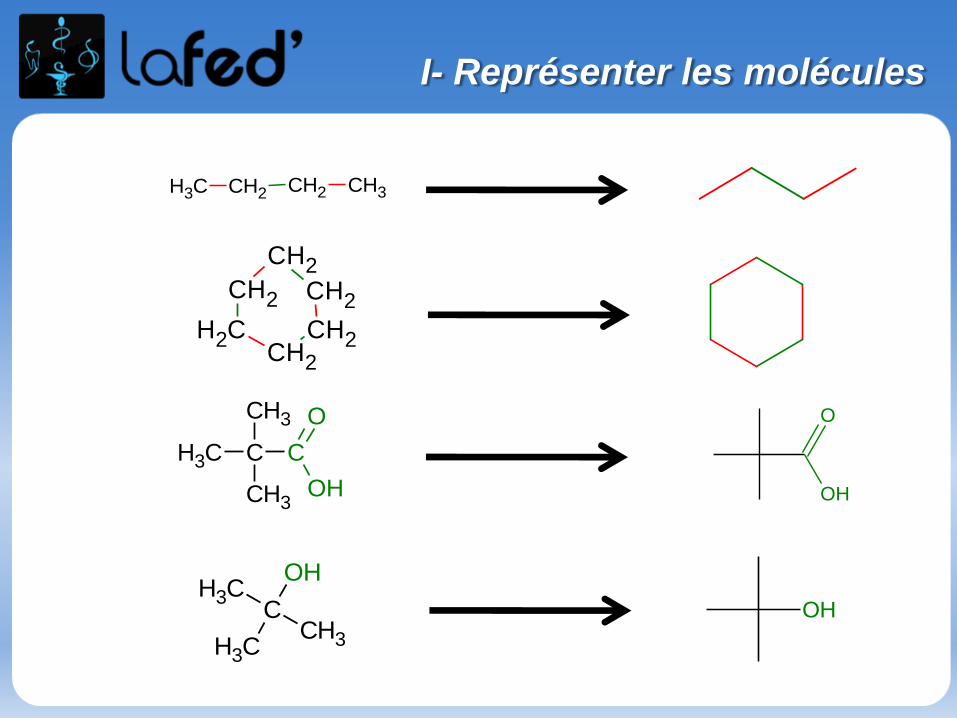
Formule topologique :
- Pas de représentation des atomes d’H, car on postule que lesatomes de carbones sont tétravalents.- Représentation des C par des points reliés entre eux par deslignes brisées.- Explicitation des fonctions chimiques ( OH, COOH… )
C CH3
H
H
HC CH2
H
H
Un carbone est « toujours » tétravalent sous forme stable ( non ionisée ).
TétravalentTétracoordonné
TétravalentTricoordonné
Remarque :
I- Représenter les molécules
CH3 CH2 CH2 CH3
CH2
CH2
CH2
CH2
CH2
CH2
CH3 C
CH3
CH3
C
O
OH
O
OH
CH3
CCH3
CH3
OH
OH
I- Représenter les molécules
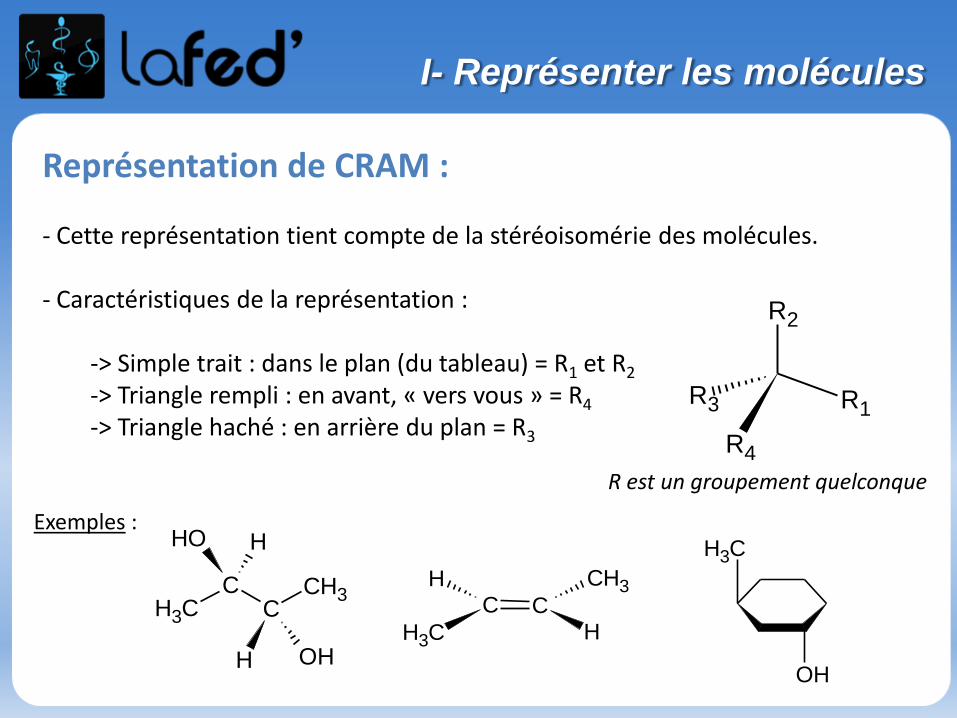
Représentation de CRAM :
- Cette représentation tient compte de la stéréoisomérie des molécules.
- Caractéristiques de la représentation :
-> Simple trait : dans le plan (du tableau) = R1 et R2
-> Triangle rempli : en avant, « vers vous » = R4
-> Triangle haché : en arrière du plan = R3 R4
R3
R2
R1
R est un groupement quelconque
CH3
CC
CH3
OH H
H OH
Exemples :
C C
CH3
CH3
H
H
OH
CH3
I- Représenter les molécules
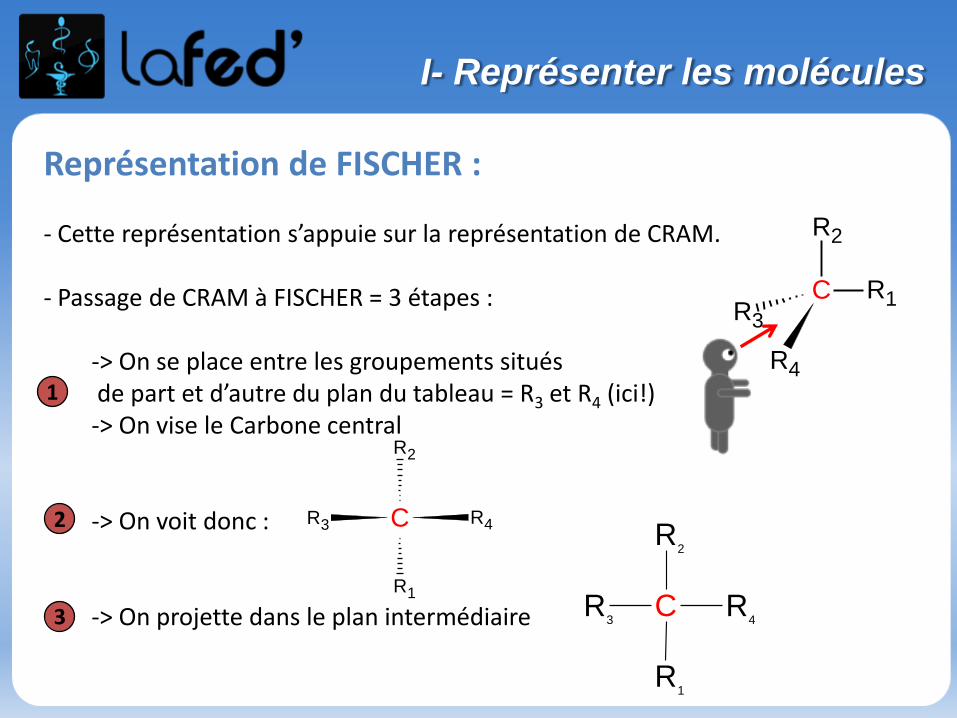
Représentation de FISCHER :
- Cette représentation s’appuie sur la représentation de CRAM.
- Passage de CRAM à FISCHER = 3 étapes :
-> On se place entre les groupements situésde part et d’autre du plan du tableau = R3 et R4 (ici!)-> On vise le Carbone central
-> On voit donc :
-> On projette dans le plan intermédiaire
C
R4
R3
R2
R1
CR3
R2
R1
R4
1
2
3 R4
R3
C
R1
R2
I- Représenter les molécules
80
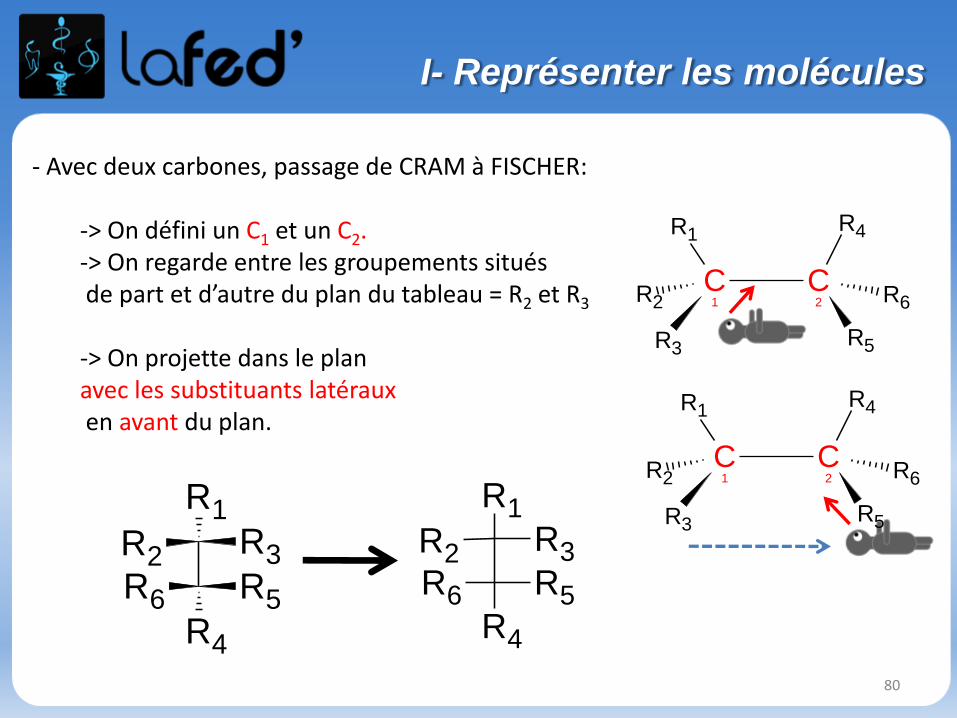
- Avec deux carbones, passage de CRAM à FISCHER:
-> On défini un C1 et un C2.-> On regarde entre les groupements situésde part et d’autre du plan du tableau = R2 et R3
-> On projette dans le plan avec les substituants latérauxen avant du plan.
C1
C2
R3
R2
R5
R6
R1R4
C1
C2
R3
R2
R5
R6
R1R4
R4
R3R2
R6 R5
R1
R4
R3R2
R6 R5
R1
I- Représenter les molécules
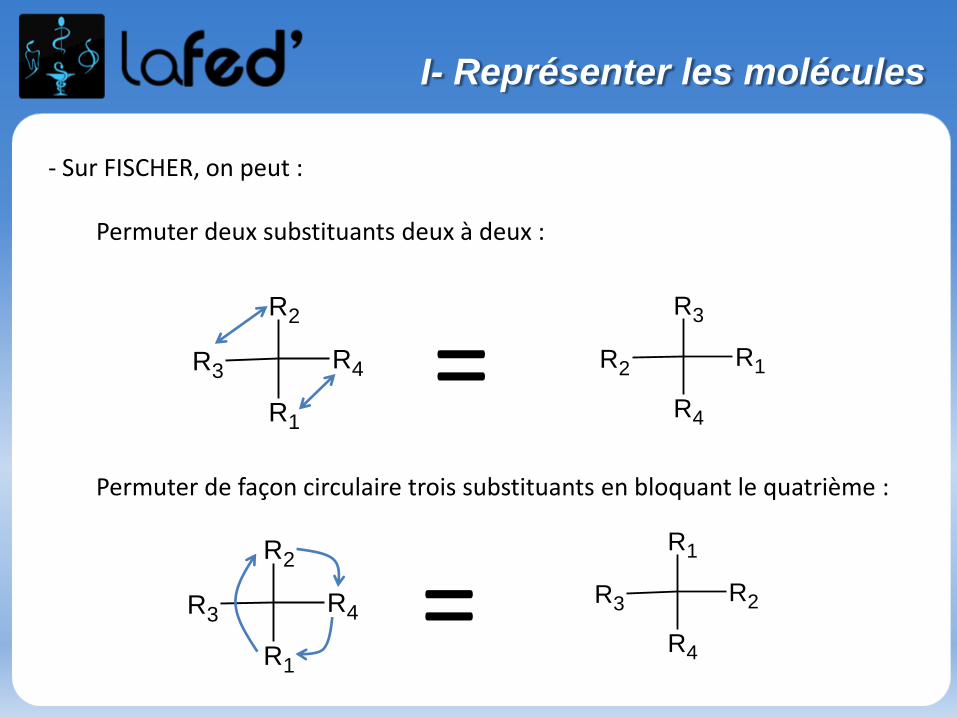
- Sur FISCHER, on peut :
Permuter deux substituants deux à deux :
Permuter de façon circulaire trois substituants en bloquant le quatrième :
=
=
R1
R4R3
R2
R4
R1R2
R3
R4
R2R3
R1
R1
R4R3
R2
I- Représenter les molécules
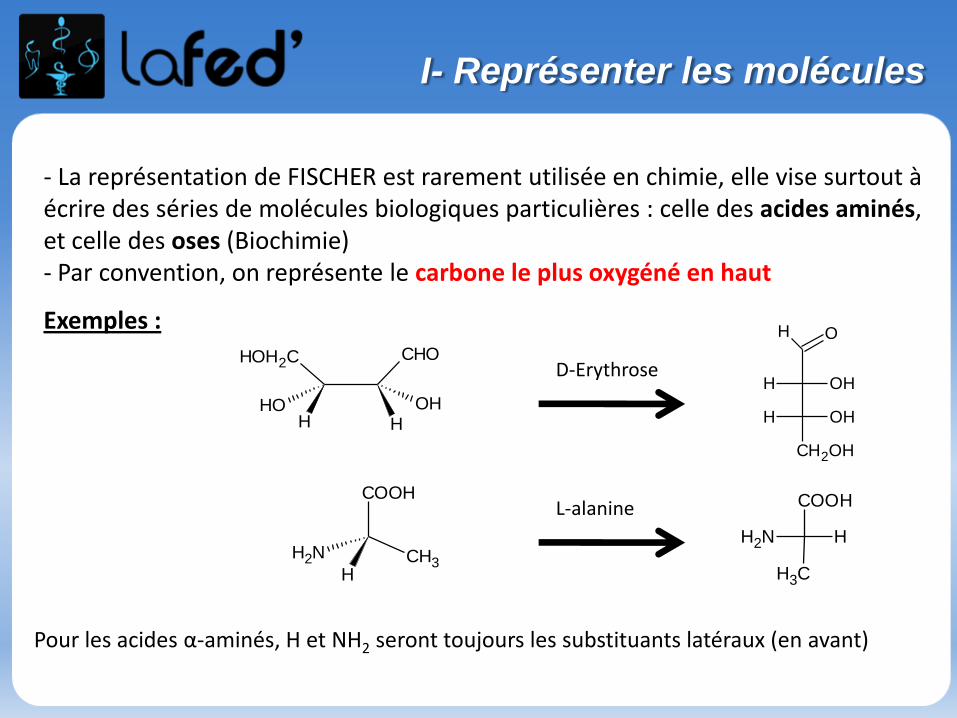
- La représentation de FISCHER est rarement utilisée en chimie, elle vise surtout àécrire des séries de molécules biologiques particulières : celle des acides aminés,et celle des oses (Biochimie)- Par convention, on représente le carbone le plus oxygéné en haut
Exemples :
HOH2C CHO
OHH H
OH
H
CH2OH
O
H OH
H OH
COOH
H
NH2 CH3
COOH
NH2 H
CH3
L-alanine
D-Erythrose
Pour les acides α-aminés, H et NH2 seront toujours les substituants latéraux (en avant)
I- Représenter les molécules
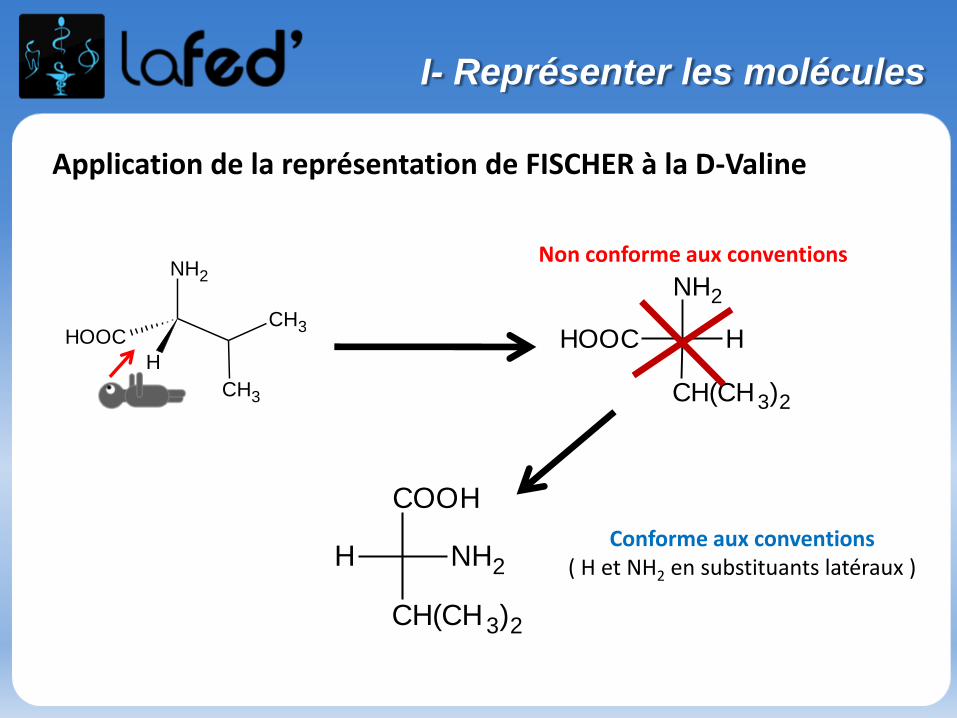
Application de la représentation de FISCHER à la D-Valine
NH2
H
HOOC
CH3
CH3
NH2
HOOC H
CH(CH3)2
COOH
H NH2
CH(CH3)2
Non conforme aux conventions
Conforme aux conventions( H et NH2 en substituants latéraux )
I- Représenter les molécules
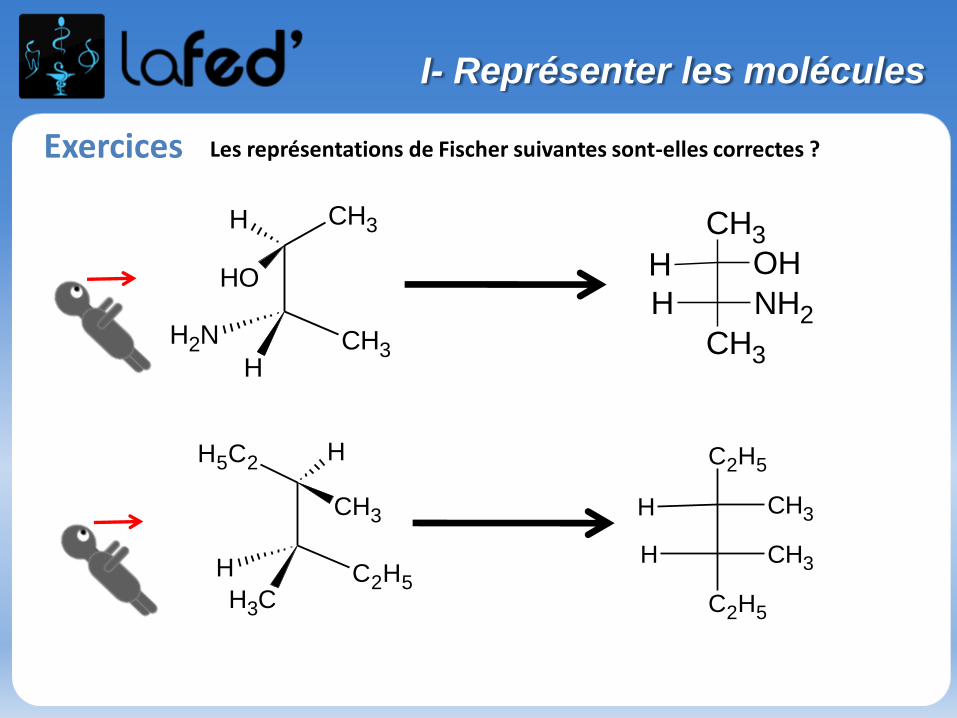
Exercices Les représentations de Fischer suivantes sont-elles correctes ?
H
NH2 CH3
CH3H
OH
CH3
OHH
H NH2
CH3
CH3
H C2H5
H5C2 H
CH3
C2H5
CH3H
H CH3
C2H5
I- Représenter les molécules
85
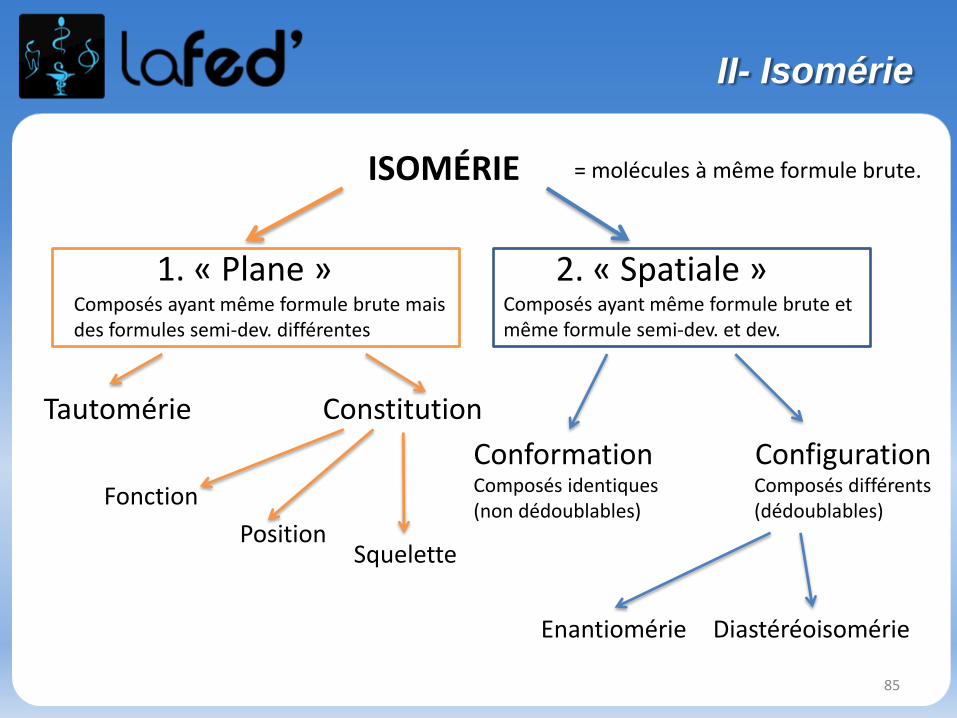
1. « Plane » 2. « Spatiale »
Tautomérie
Fonction
Position
Conformation ConfigurationComposés identiques(non dédoublables)
Composés différents(dédoublables)
Composés ayant même formule brute mais des formules semi-dev. différentes
Composés ayant même formule brute et même formule semi-dev. et dev.
Enantiomérie
Constitution
Diastéréoisomérie
Squelette
ISOMÉRIE = molécules à même formule brute.
II- Isomérie
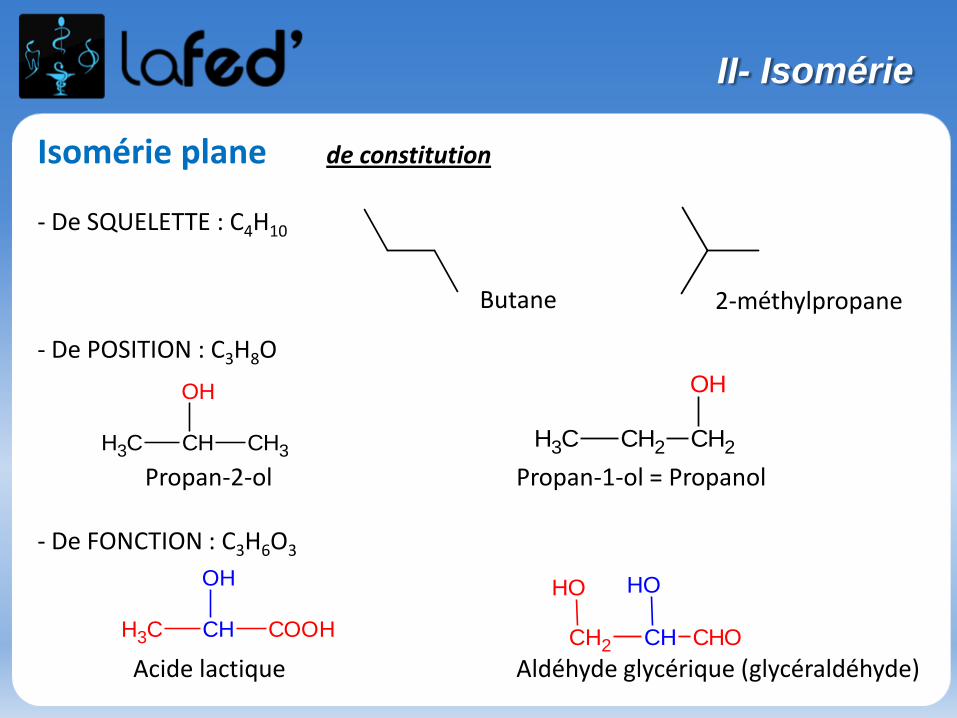
Isomérie plane de constitution
- De SQUELETTE : C4H10
- De POSITION : C3H8O
Propan-2-ol Propan-1-ol = Propanol
- De FONCTION : C3H6O3
Acide lactique Aldéhyde glycérique (glycéraldéhyde)
CH3 CH COOH
OH
CH2 CH CHO
OHOH
CH3 CH CH3
OH
CH3 CH2 CH2
OH
Butane 2-méthylpropane
II- Isomérie
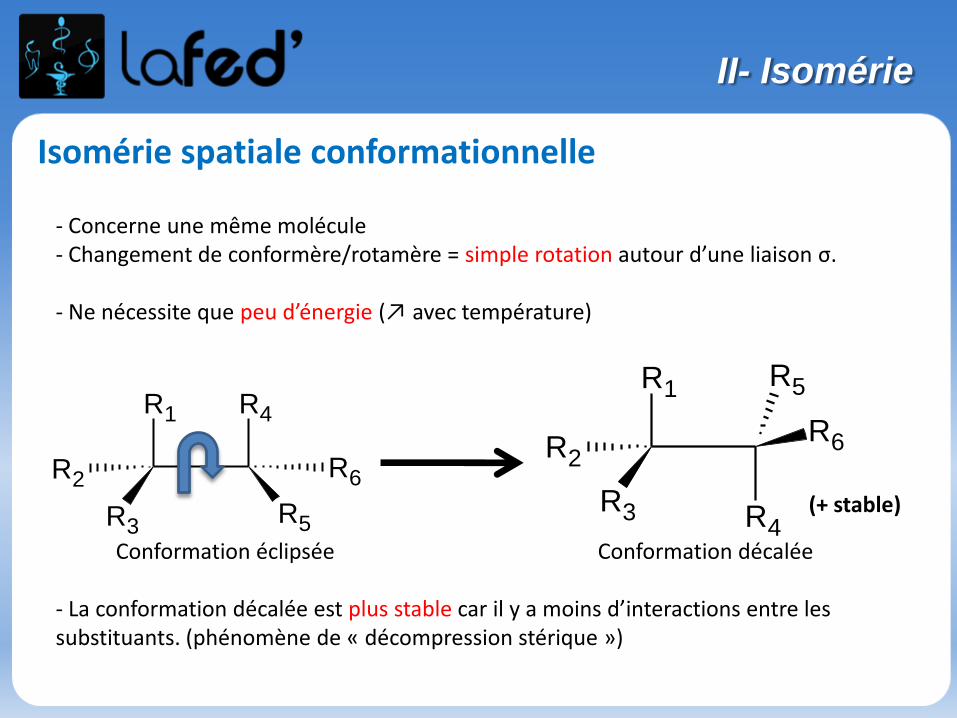
Isomérie spatiale conformationnelle
- Concerne une même molécule- Changement de conformère/rotamère = simple rotation autour d’une liaison σ.
- Ne nécessite que peu d’énergie (↗ avec température)
Conformation éclipsée Conformation décalée
- La conformation décalée est plus stable car il y a moins d’interactions entre les substituants. (phénomène de « décompression stérique »)
R3R5
R6R2
R1 R4
R3
R5
R6R2
R1
R4(+ stable)
II- Isomérie

-Concerne deux composés différents = dédoublables.
- On distingue deux types d’isomérie configurationnelle : a) L’énantiomérieb) La diastéréoisomérie
- Ces deux notions reposent assez largement sur le concept de « carboneasymétrique » (noté C*) = carbone ayant 4 substituants différents.
- Dans un premier temps nous étudierons les règles de nomenclature associéesà ces carbones asymétriques.
Isomérie spatiale configurationnelle
II- Isomérie

Nomenclature R/S
- On détermine la configuration absolue d’un C* à partir d’un classement de ses substituants.
- Classement basé sur les numéros atomiques (Z) des atomes liés au C*(Convention de « Cahn, Ingold et Prelog »)
ZO = 8 > ZN = 7 > Zc = 6 > ZH = 1 > Zd.n.l = 0
• Remarque :-Dans le cas d’isotopes (de l’hydrogène souvent : deutérium D…), on classe par masseatomique décroissante.T > D > H
II- Isomérie
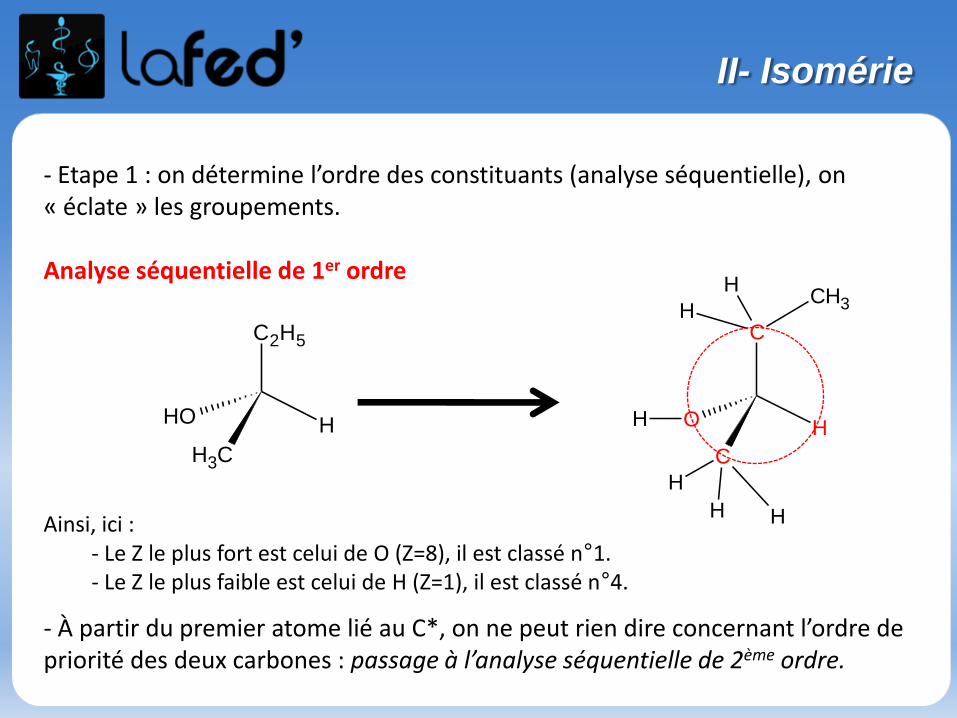
- Etape 1 : on détermine l’ordre des constituants (analyse séquentielle), on « éclate » les groupements.
Analyse séquentielle de 1er ordre
Ainsi, ici :- Le Z le plus fort est celui de O (Z=8), il est classé n°1.- Le Z le plus faible est celui de H (Z=1), il est classé n°4.
- À partir du premier atome lié au C*, on ne peut rien dire concernant l’ordre de priorité des deux carbones : passage à l’analyse séquentielle de 2ème ordre.
C2H5
HOH
CH3
H
C
HO
C
CH3H
H
H H
H
II- Isomérie
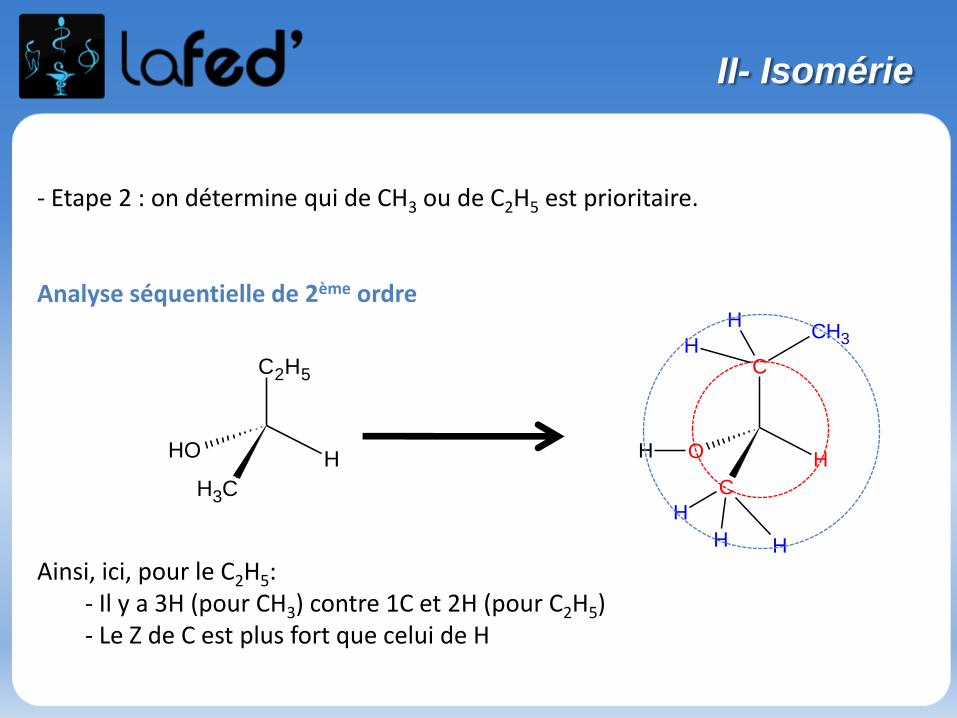
- Etape 2 : on détermine qui de CH3 ou de C2H5 est prioritaire.
Analyse séquentielle de 2ème ordre
Ainsi, ici, pour le C2H5:- Il y a 3H (pour CH3) contre 1C et 2H (pour C2H5)- Le Z de C est plus fort que celui de H
C2H5
HOH
CH3
H
C
HO
C
CH3H
H
H H
H
II- Isomérie
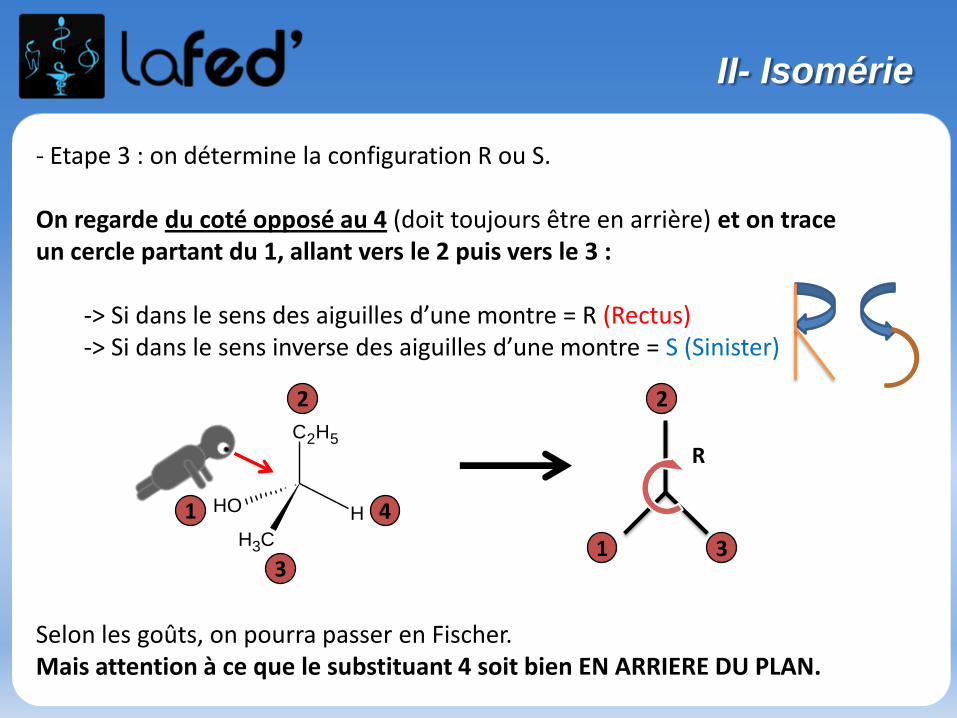
- Etape 3 : on détermine la configuration R ou S.
On regarde du coté opposé au 4 (doit toujours être en arrière) et on trace un cercle partant du 1, allant vers le 2 puis vers le 3 :
-> Si dans le sens des aiguilles d’une montre = R (Rectus)-> Si dans le sens inverse des aiguilles d’une montre = S (Sinister)
Selon les goûts, on pourra passer en Fischer.Mais attention à ce que le substituant 4 soit bien EN ARRIERE DU PLAN.
C2H5
HOH
CH3
1
2
4
31
2
R
3
II- Isomérie
1
2
34
R
1
2
3
4
COOH
HCH2OH
NH2
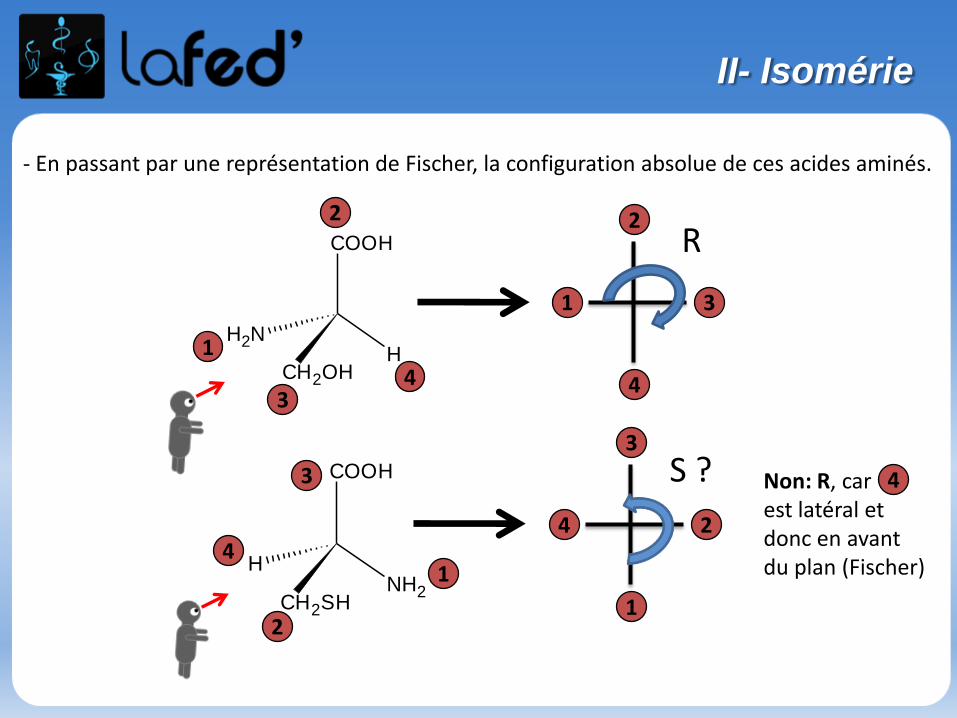
- En passant par une représentation de Fischer, la configuration absolue de ces acides aminés.
COOH
NH2CH2SH
H 1
2
3
44
3
2
1
S ? Non: R, carest latéral et donc en avantdu plan (Fischer)
4
II- Isomérie
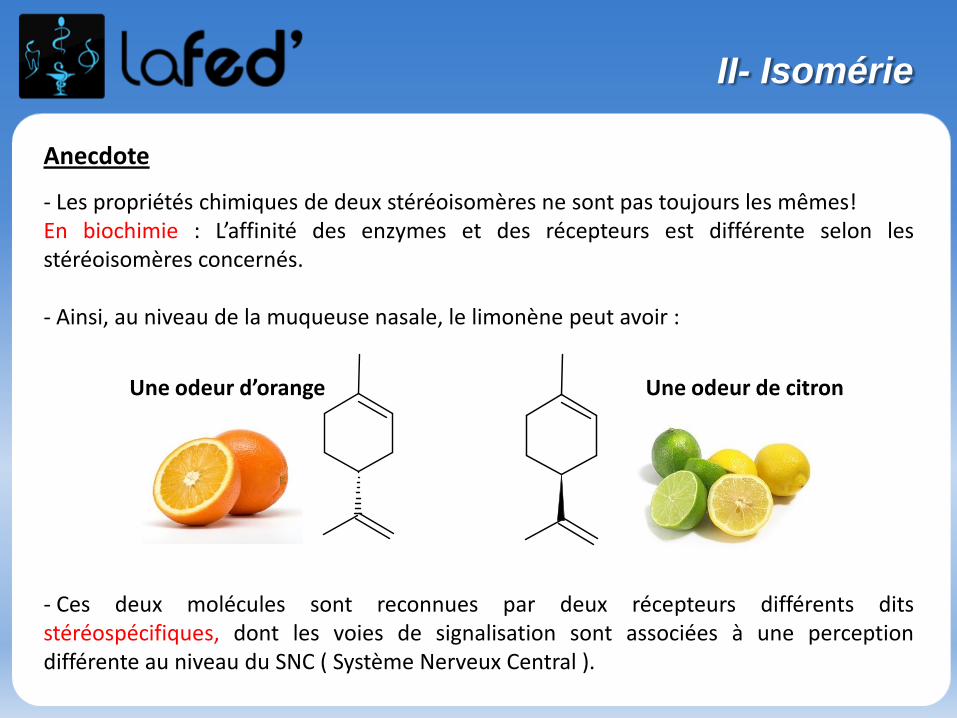
Anecdote
- Les propriétés chimiques de deux stéréoisomères ne sont pas toujours les mêmes!En biochimie : L’affinité des enzymes et des récepteurs est différente selon lesstéréoisomères concernés.
- Ainsi, au niveau de la muqueuse nasale, le limonène peut avoir :
- Ces deux molécules sont reconnues par deux récepteurs différents ditsstéréospécifiques, dont les voies de signalisation sont associées à une perceptiondifférente au niveau du SNC ( Système Nerveux Central ).
Une odeur de citronUne odeur d’orange
II- Isomérie

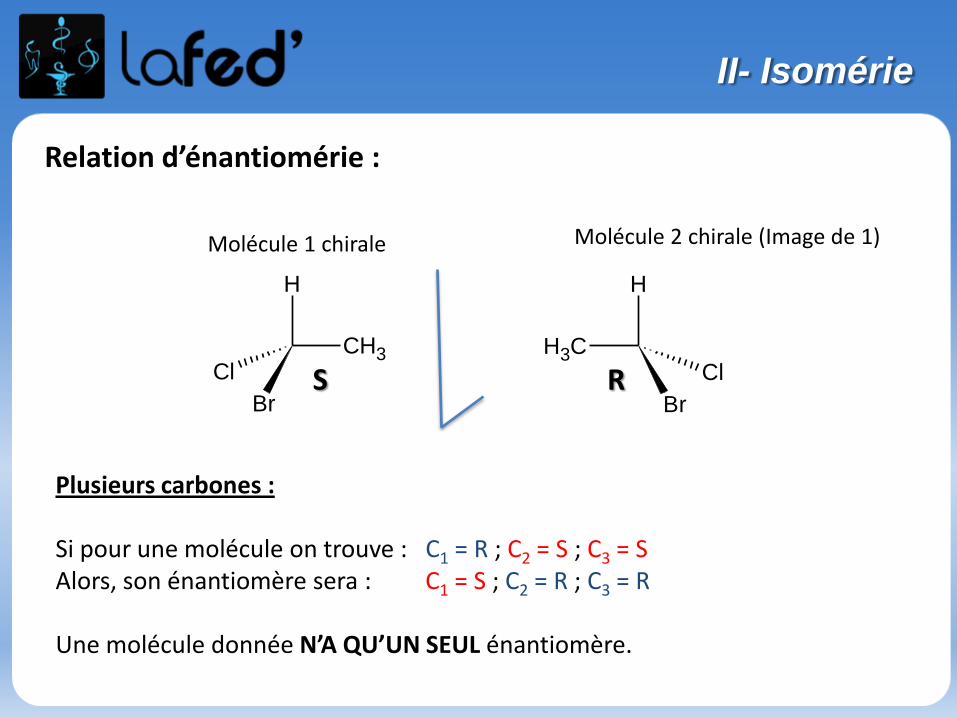
Enantiomérie :
Isomérie optique, spécifique des molécules chirales
- Une molécule chirale :-> n’a ni plan de symétrie, ni centre de symétrie-> n’est pas superposable à son image dans un miroir
(exemple de la main droite et de la main gauche).
Sinon, on parle de molécule achirale.
Une molécule chirale possède souvent 1 carbone asymétrique (noté C*).
→ Une molécule possédant 1 C*est forcément chirale.→ Une molécule possédant 2 ou plusieurs C* de symétrie est souventchirale, mais pas toujours.
II- Isomérie
Br
Cl
H
CH3
S
Molécule 1 chirale
Br
Cl
H
CH3
R
Molécule 2 chirale (Image de 1)
Relation d’énantiomérie :
Plusieurs carbones :
Si pour une molécule on trouve : C1 = R ; C2 = S ; C3 = S Alors, son énantiomère sera : C1 = S ; C2 = R ; C3 = R
Une molécule donnée N’A QU’UN SEUL énantiomère.
II- Isomérie
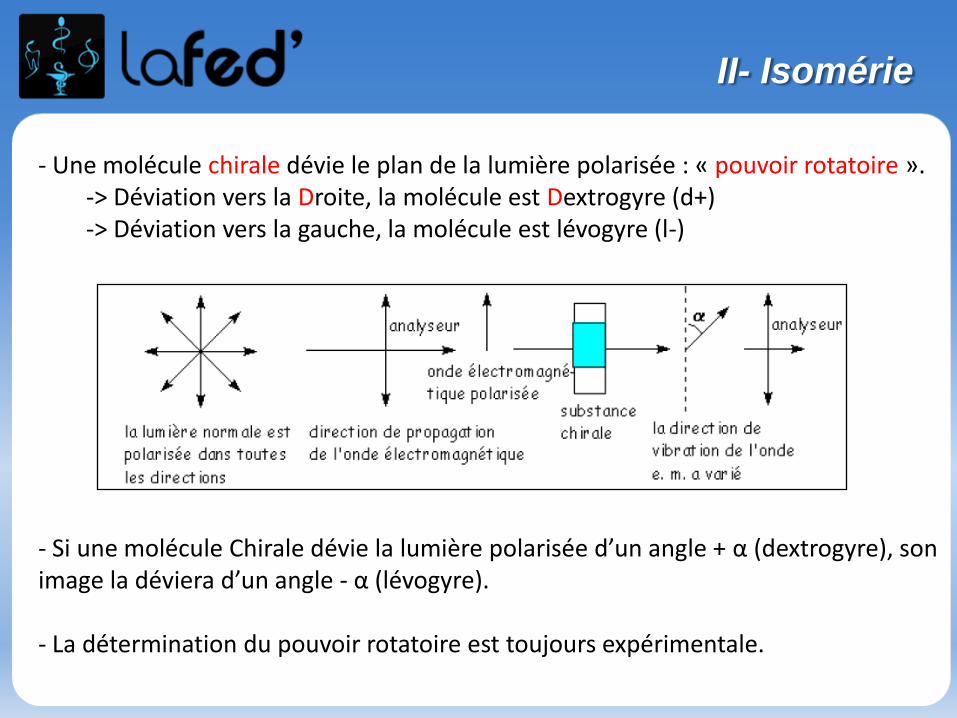
- Une molécule chirale dévie le plan de la lumière polarisée : « pouvoir rotatoire ».-> Déviation vers la Droite, la molécule est Dextrogyre (d+)-> Déviation vers la gauche, la molécule est lévogyre (l-)
- Si une molécule Chirale dévie la lumière polarisée d’un angle + α (dextrogyre), son image la déviera d’un angle - α (lévogyre).
- La détermination du pouvoir rotatoire est toujours expérimentale.
II- Isomérie

Diastéréoisomères = Tout couple de stéréoisomères n’étant pas liés par unerelation d’énantiomérie.
Plusieurs carbones :
Si pour une molécule on trouve : C1 = R ; C2 = S ; C3 = S
Alors, ses diastéréoisomères seront : C1 = S ; C2 = S ; C3 = SC1 = R ; C2 = R ; C3 = RC1 = R ; C2 = R ; C3 = SC1 = R ; C2 = S ; C3 = R C1 = S ; C2 = R ; C3 = S C1 = S ; C2 = S ; C3 = R
Diastéréoisomérie
II- Isomérie
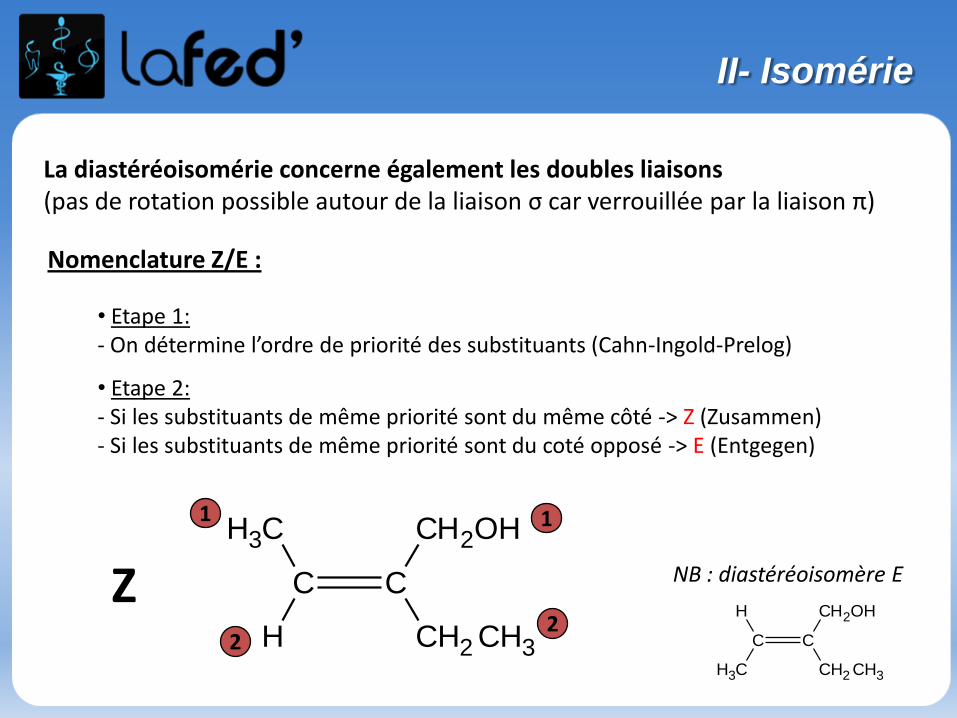
La diastéréoisomérie concerne également les doubles liaisons(pas de rotation possible autour de la liaison σ car verrouillée par la liaison π)
Nomenclature Z/E :
• Etape 1:- On détermine l’ordre de priorité des substituants (Cahn-Ingold-Prelog)
• Etape 2:- Si les substituants de même priorité sont du même côté -> Z (Zusammen)- Si les substituants de même priorité sont du coté opposé -> E (Entgegen)
CH3
C
H
C
CH2OH
CH2 CH3
1
2
1
2Z NB : diastéréoisomère E
H
C
CH3
C
CH2OH
CH2 CH3
II- Isomérie
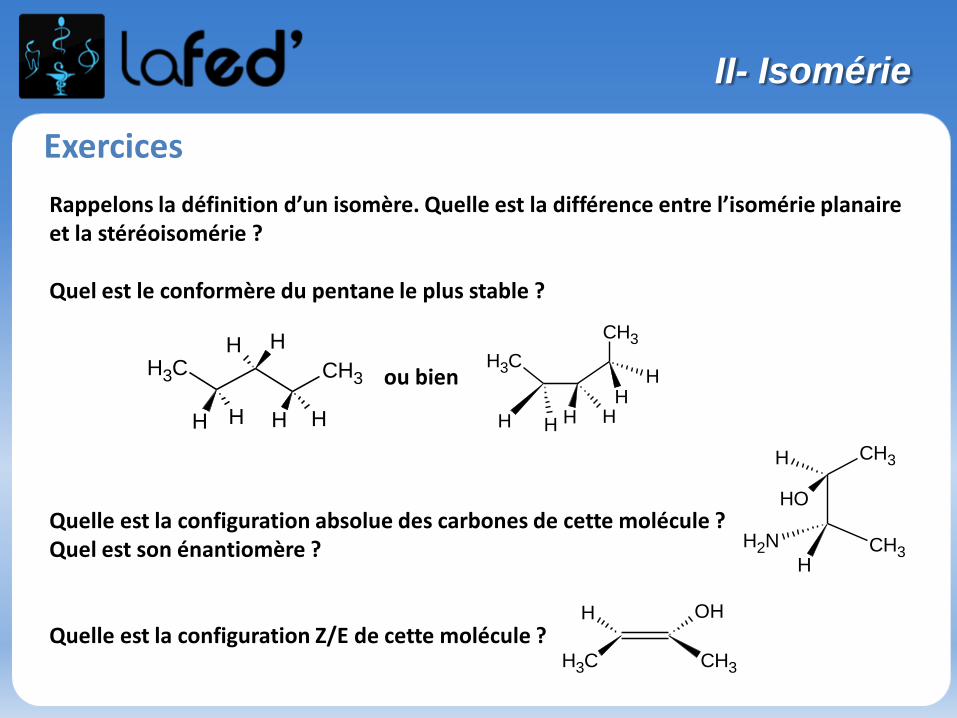
Exercices
Rappelons la définition d’un isomère. Quelle est la différence entre l’isomérie planaire et la stéréoisomérie ?
Quel est le conformère du pentane le plus stable ?
ou bien
Quelle est la configuration absolue des carbones de cette molécule ?Quel est son énantiomère ?
Quelle est la configuration Z/E de cette molécule ?
II- Isomérie
CH3 CH3
H
H
HH
H
H
CH3
CH3
H HH
H H
H
H
NH2 CH3
CH3H
OH
OHH
CH3 CH3

Stage de Pré-Rentrée 2012UE 1 – Effets électroniques
101
Diaporama réalisé par les tuteurs de La Fed’

102
• Des effets électroniques peuvent être responsables de la polarisation(permanente ou instantanée d’une liaison)
• Ces effets sont d’une importance cruciale pour comprendre et étudier lesmécanismes des réactions chimiques.
•Les effets inductifs et mésomères peuvent co-exister au sein d’une mêmemolécule, mais l’effet mésomère (s’il existe) prédominera souvent sur l’effetinductif.
Effet mésomère > Effet inductif
Généralités

Effet inductif statique
- Définition : Effet de polarisation permanente des liaisons σ par des atomes ou groupement d’atomes. L’effet inductif ne concerne que les e- des liaisons σ.
- Rappel : les 2 électrons d’une liaison occupent l’ensemble du volume de l’orbitale moléculaire sans que l’on puisse leur affecter une position précise à un instant donné. On ne leur attribue qu’une probabilité de présence.
Si les 2 atomes sont différents, la probabilité de présence est dissymétrique.
+δ -δ
(χ = 3,1) (χ=3,5)
- (µ ≠ 0)- Au niveau de cette liaison σ, le doublet électronique aura plus tendance à aller vers le Cl
I- Effet inductif
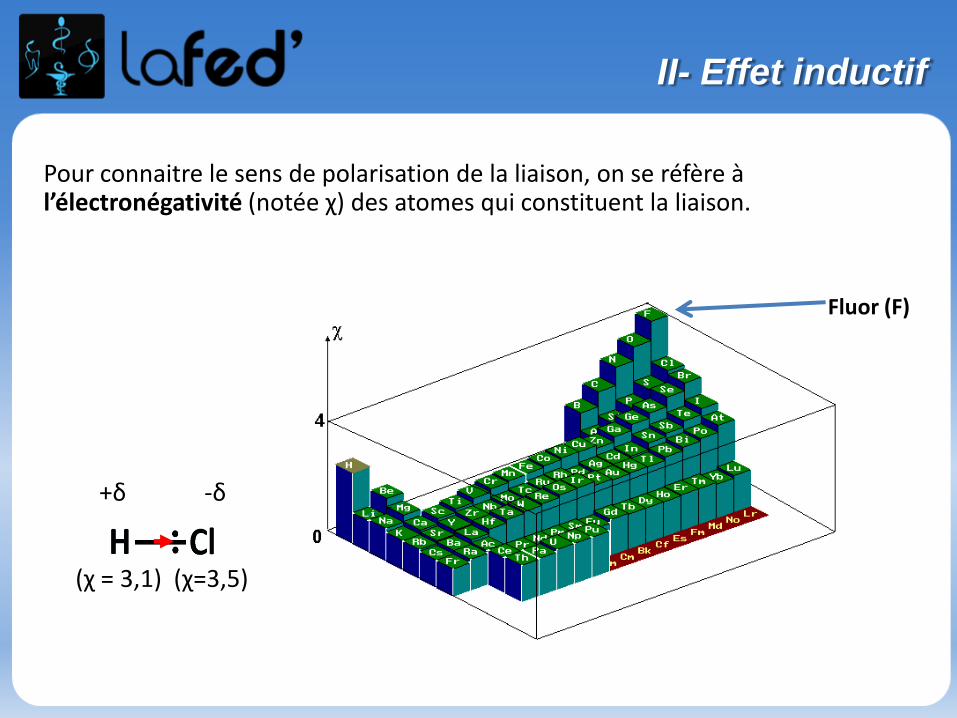
Pour connaitre le sens de polarisation de la liaison, on se réfère à l’électronégativité (notée χ) des atomes qui constituent la liaison.
Fluor (F)
+δ -δ
(χ = 3,1) (χ=3,5)
II- Effet inductif
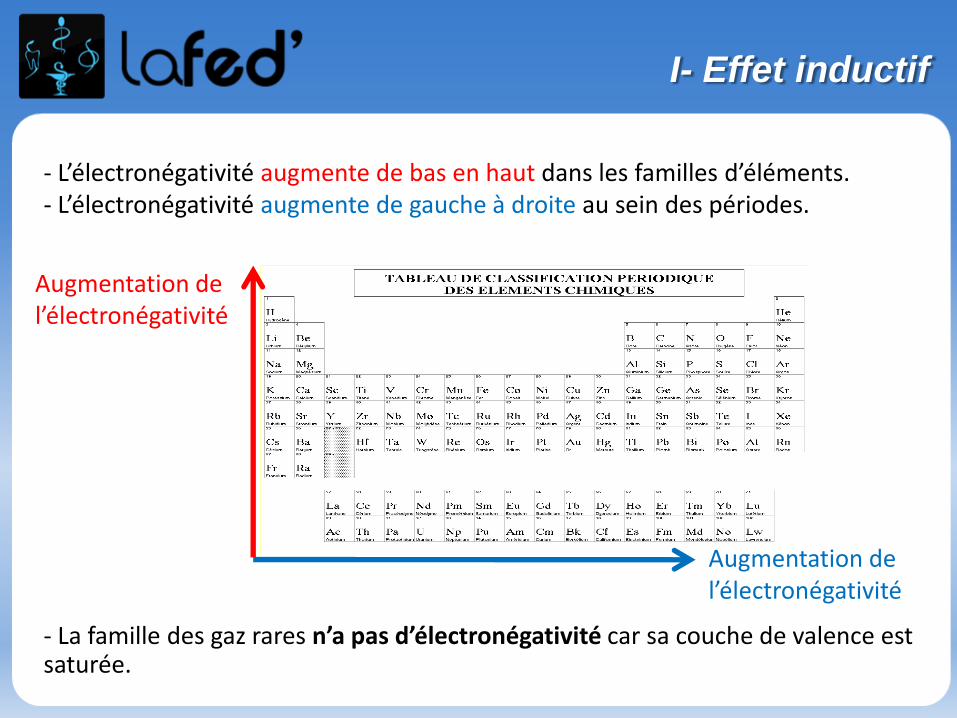
- L’électronégativité augmente de bas en haut dans les familles d’éléments.- L’électronégativité augmente de gauche à droite au sein des périodes.
- La famille des gaz rares n’a pas d’électronégativité car sa couche de valence est saturée.
Augmentation de l’électronégativité
Augmentation de l’électronégativité
I- Effet inductif

106
- L’électronégativité inclut la propriété d’un atome à attirer vers lui les électrons des liaisons établies avec d’autres atomes (= « affinité électronique »).
- On attribue pour chaque élément de la classification périodique, une valeur (χ) représentative du caractère électronégatif de cet atome.
Exemple : Soit la liaison A B (avec B plus électronégatif que A).
- On note les charges partielles : + δ et - δ
+δ -δ
I- Effet inductif
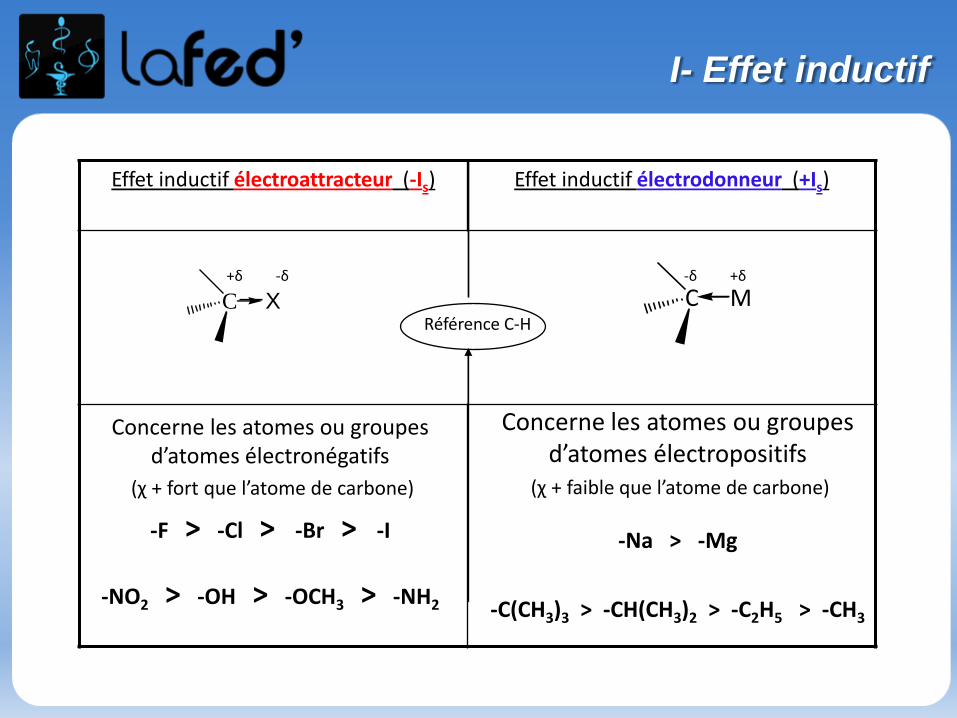
Effet inductif électroattracteur (-Is) Effet inductif électrodonneur (+Is)
+δ -δ -δ +δ
Référence C-H C X C M
Concerne les atomes ou groupes d’atomes électronégatifs
(χ + fort que l’atome de carbone)
-F > -Cl > -Br > -I
-NO2 > -OH > -OCH3 > -NH2
Concerne les atomes ou groupes d’atomes électropositifs
(χ + faible que l’atome de carbone)
-Na > -Mg
-C(CH3)3 > -CH(CH3)2 > -C2H5 > -CH3
I- Effet inductif
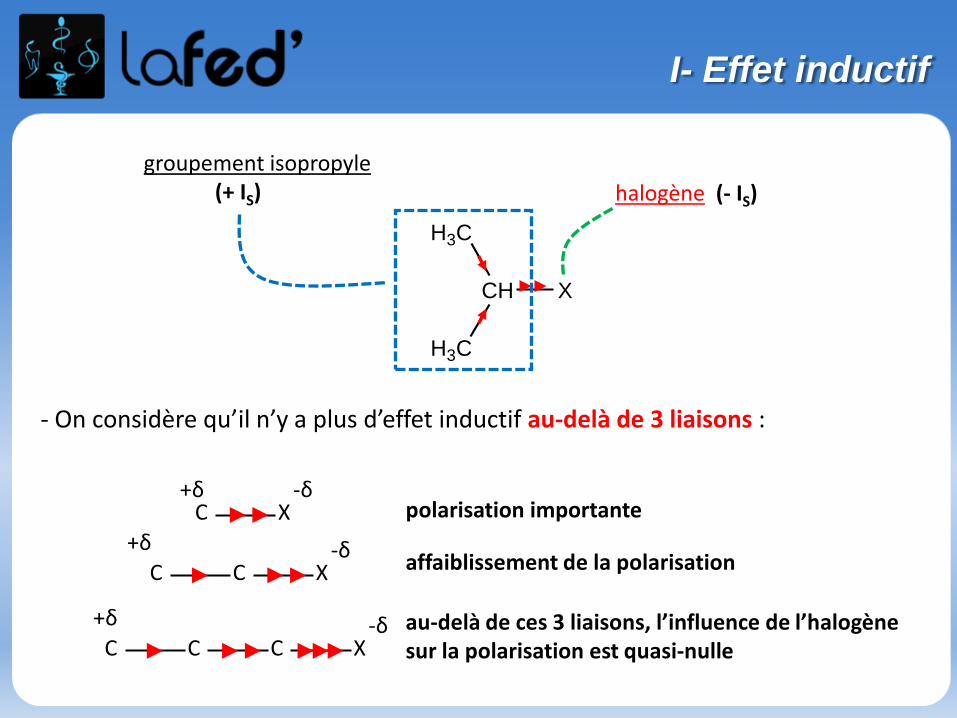
- On considère qu’il n’y a plus d’effet inductif au-delà de 3 liaisons :
H3C
CH
H3C
X
C X-δ+δ
C X-δ+δ
C
C X-δ+δ
CC
polarisation importante
affaiblissement de la polarisation
au-delà de ces 3 liaisons, l’influence de l’halogènesur la polarisation est quasi-nulle
groupement isopropyle(+ IS) halogène (- IS)
I- Effet inductif

Conséquences : Moment dipolaire
- Une grandeur directement liée à ce phénomène est le moment dipolaire noté « μ ».
a) Dans une molécule symétrique : les électrons participantaux liaisons sont répartis également entre les atomesLes centres de gravité des charges positives et négatives coïncident .
b) Dans une molécule asymétrique : un des atomes retient lamajeure partie des électrons, il apparaît un dipôle composé desnoyaux porteurs chacun de la différence de charges à chaqueextrémités
- Ce moment dipolaire a une unité : le DEBYE
μ = q x r
C C
H
Br
Br
H
alcène E(μ = 0)Centre de symétrie
C C
H
Br
H
Br
alcène Z(μ ≠ 0)Répartition des charges
q : charge d’un e-
r : longueur de la liaison(1D = 3,336 .10-30 C.m)
I- Effet inductif
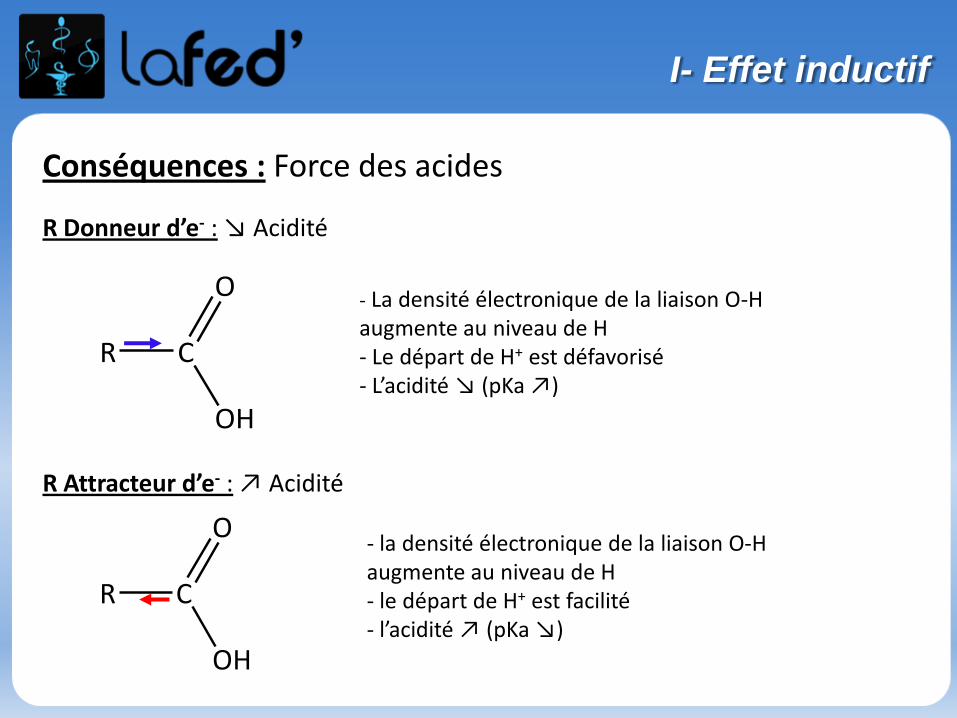
Conséquences : Force des acides
R Donneur d’e- : ↘ Acidité
R Attracteur d’e- : ↗ Acidité
- La densité électronique de la liaison O-H augmente au niveau de H- Le départ de H+ est défavorisé- L’acidité ↘ (pKa ↗)
- la densité électronique de la liaison O-H augmente au niveau de H- le départ de H+ est facilité- l’acidité ↗ (pKa ↘)
R C
O
OH
R C
O
OH
I- Effet inductif

111
Effet inductif dynamique (ID)
- Cette polarisation est temporaire : notion de polarisabilité de la liaison.
- La polarisabilité d’une liaison = son aptitude à se polariser à l’approche d’un réactif (déformation des orbitales)
- La polarisabilité :- dépend du rayon atomique - est inversement proportionnelle à la polarisation de la liaison au départ
C-Cl est moins polarisable que C-I car I est plus volumineux que Cl→ Ainsi, la liaison sera plus facilement "déformée" a proximité d'un réactif.
X X
Nu-
Polarité C-F > C-Cl > C-Br > C-I
Polarisabilité C-F < C-Cl < C-Br < C-I
+
+
-
-
I- Effet inductif
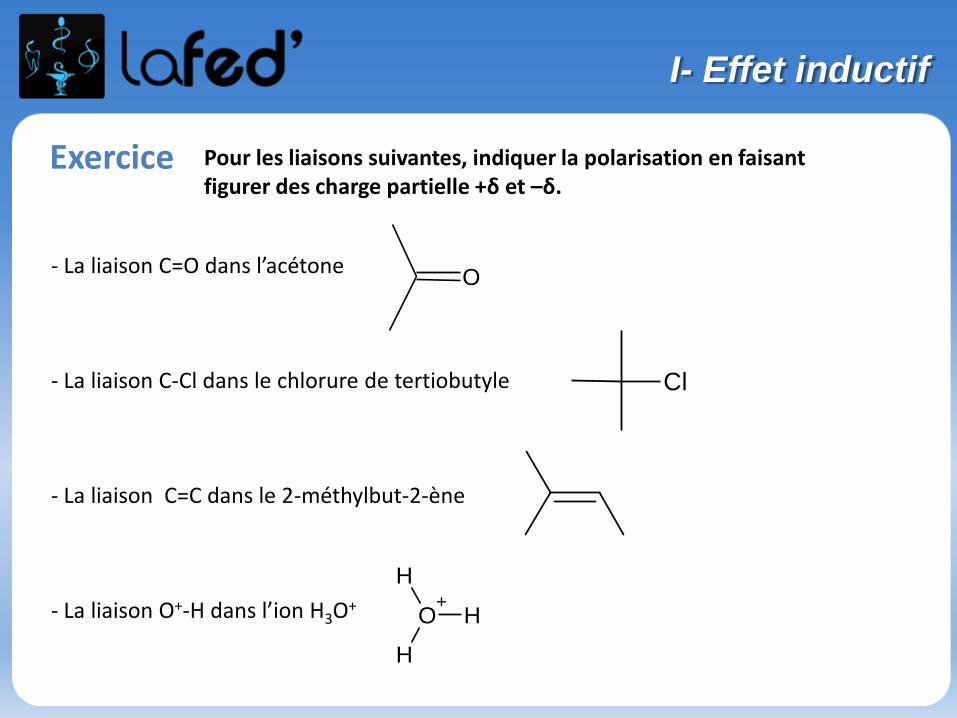
Exercice Pour les liaisons suivantes, indiquer la polarisation en faisant figurer des charge partielle +δ et –δ.
- La liaison C=O dans l’acétone
- La liaison C-Cl dans le chlorure de tertiobutyle
- La liaison C=C dans le 2-méthylbut-2-ène
- La liaison O+-H dans l’ion H3O+
Cl
O
O+
H
H
H
I- Effet inductif
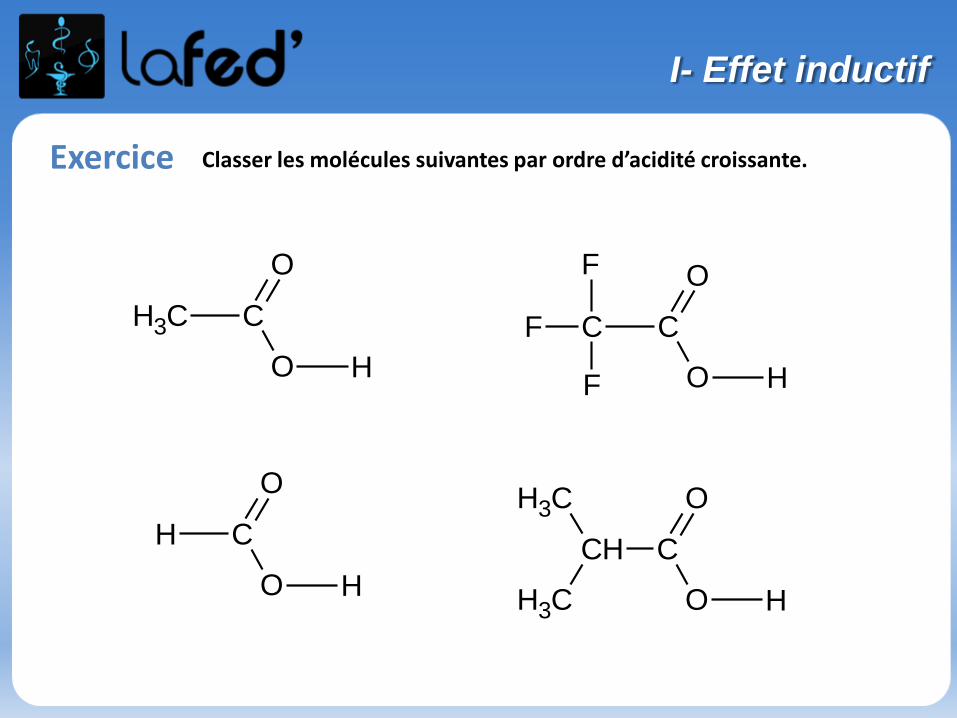
Exercice Classer les molécules suivantes par ordre d’acidité croissante.
CH C
O
O
H
CH3
CH3
C C
O
O
H
F
F
F
H C
O
O
H
CH3 C
O
O
H
I- Effet inductif
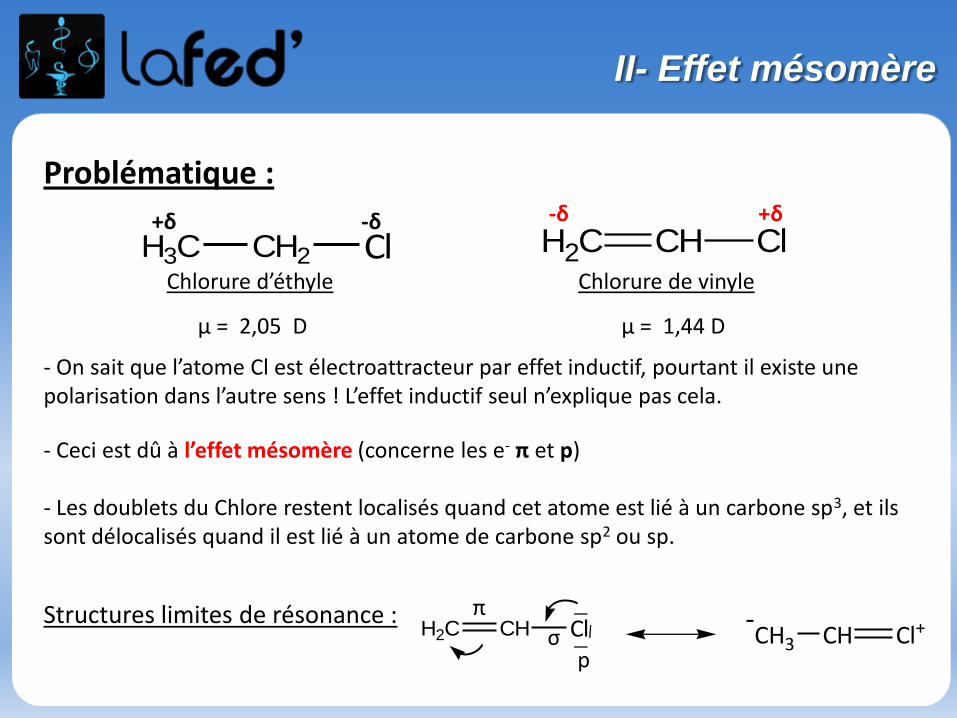
Problématique :
- On sait que l’atome Cl est électroattracteur par effet inductif, pourtant il existe une polarisation dans l’autre sens ! L’effet inductif seul n’explique pas cela.
- Ceci est dû à l’effet mésomère (concerne les e- π et p)
- Les doublets du Chlore restent localisés quand cet atome est lié à un carbone sp3, et ils sont délocalisés quand il est lié à un atome de carbone sp2 ou sp.
Structures limites de résonance :
H3C CH2 Cl
H2C CH Cl_
__ CH3 CH Cl+-
Chlorure d’éthyle
μ = 2,05 D
Chlorure de vinyle
μ = 1,44 D
+δ -δ
p
π
σ
CH2 CH Cl-δ +δ
II- Effet mésomère
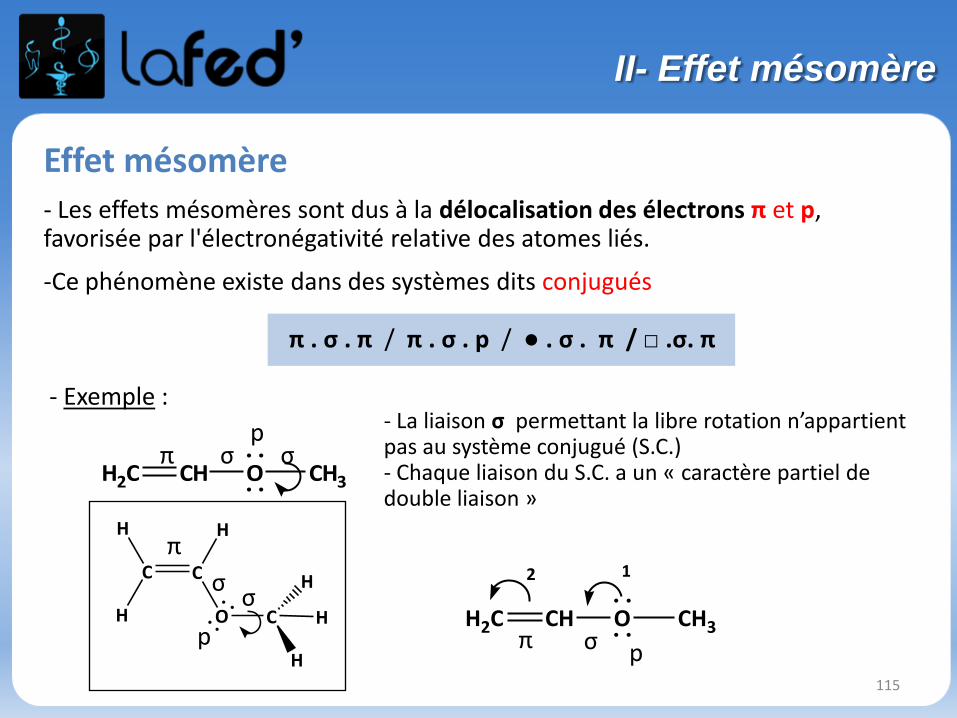
115
Effet mésomère- Les effets mésomères sont dus à la délocalisation des électrons π et p, favorisée par l'électronégativité relative des atomes liés.
-Ce phénomène existe dans des systèmes dits conjugués
π . σ . π / π . σ . p / ● . σ . π / □ .σ. π
- Exemple :
. .
H
C C
H
H
O C H
H
H
. .
H2C CH O CH3
. .
. .
π
σσ
π σp
σ
pH2C CH O CH3
. .
. .
12
- La liaison σ permettant la libre rotation n’appartientpas au système conjugué (S.C.) - Chaque liaison du S.C. a un « caractère partiel de double liaison »
pσπ
II- Effet mésomère
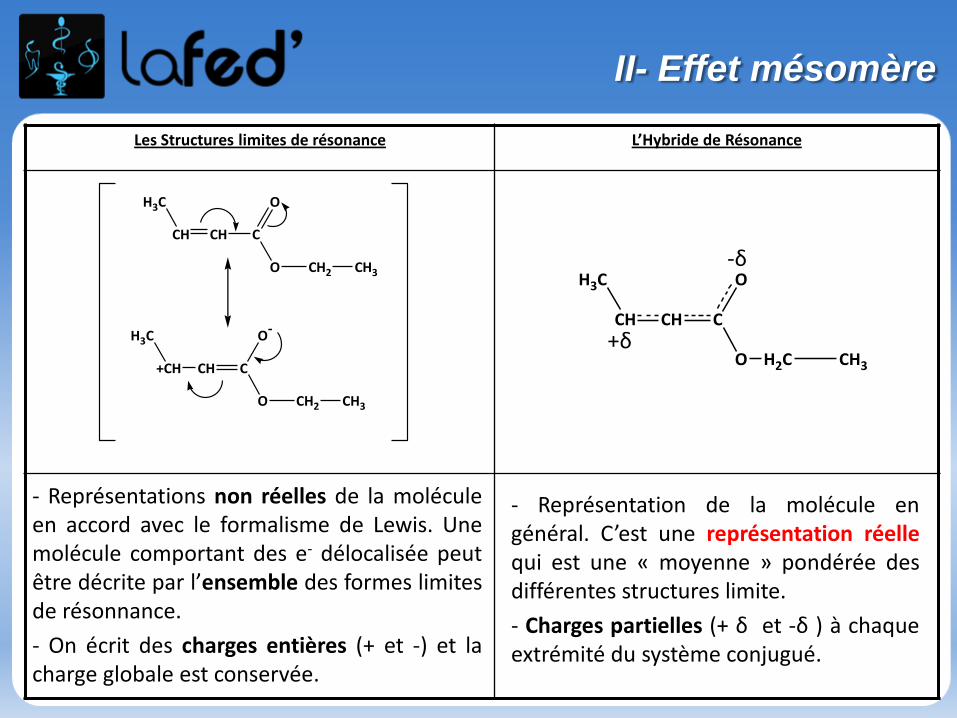
Les Structures limites de résonance L’Hybride de Résonance
H3C
CH CH C
O
O CH2 CH3
H3C
+CH CH C
O-
O CH2 CH3
H3C
CH CH C
O
O H2C CH3
-δ
+δ
- Représentations non réelles de la moléculeen accord avec le formalisme de Lewis. Unemolécule comportant des e- délocalisée peutêtre décrite par l’ensemble des formes limitesde résonnance.
- On écrit des charges entières (+ et -) et lacharge globale est conservée.
- Représentation de la molécule engénéral. C’est une représentation réellequi est une « moyenne » pondérée desdifférentes structures limite.
- Charges partielles (+ δ et -δ ) à chaqueextrémité du système conjugué.
II- Effet mésomère
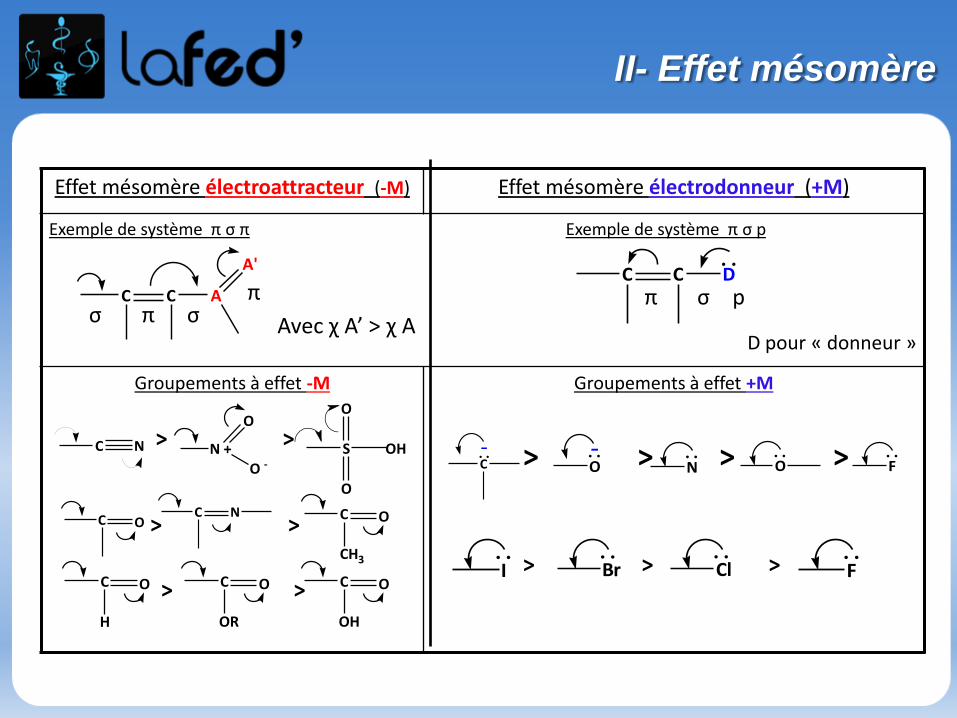
> > > >
> > >
Effet mésomère électroattracteur (-M) Effet mésomère électrodonneur (+M)
Exemple de système π σ π Exemple de système π σ p
Groupements à effet -M Groupements à effet +M
. .C C D
σπ p
D pour « donneur »
C C A
A'
π σπ
σ
C N N +
O
O -S
O
O
OH
C OC N C
CH3
O
C
H
O C
OR
O C
OH
O
-C. . . .
O- . .
N. .O
. .F
. .F
. .Cl
. .Br
. .I
> >
> >
> >
II- Effet mésomère
Avec χ A’ > χ A

Conséquences :
Caractéristiques des liaisons d’un système conjugué (S.C.) :-> Elles ont un « caractère partiel de double liaison » :-> Tendance à l’harmonisation de la longueur des liaisons :
Liaison double : ↗ de la longueur de la liaisonLiaison simple : ↘ de la longueur de la liaison
- Pas de libre rotation de la liaison σ, car le caractère partiel « liaison π » l’empêche
- Planéité des S.C., sans qu’elle ne concerne forcément toute la molécule
- La stabilité de la molécule augmente +++ grâce à l’énergie de résonance
Délocalisation des électrons = STABILISATION
II- Effet mésomère
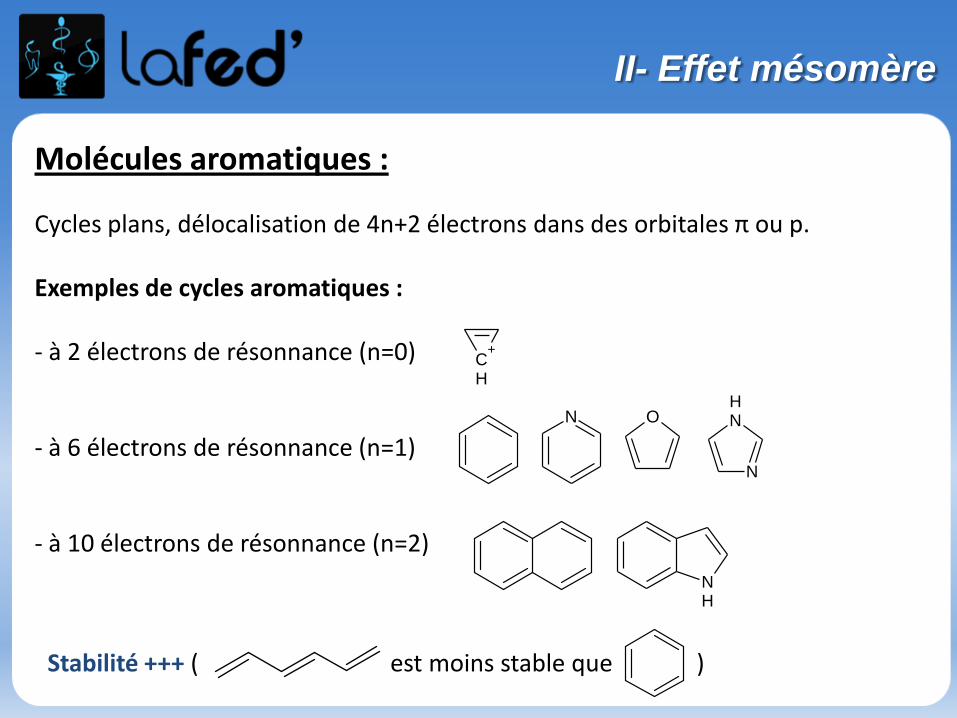
Molécules aromatiques :
Cycles plans, délocalisation de 4n+2 électrons dans des orbitales π ou p.
Exemples de cycles aromatiques :
- à 2 électrons de résonnance (n=0)
- à 6 électrons de résonnance (n=1)
- à 10 électrons de résonnance (n=2)
CH
+
N O NH
N
NH
Stabilité +++ ( est moins stable que )
II- Effet mésomère
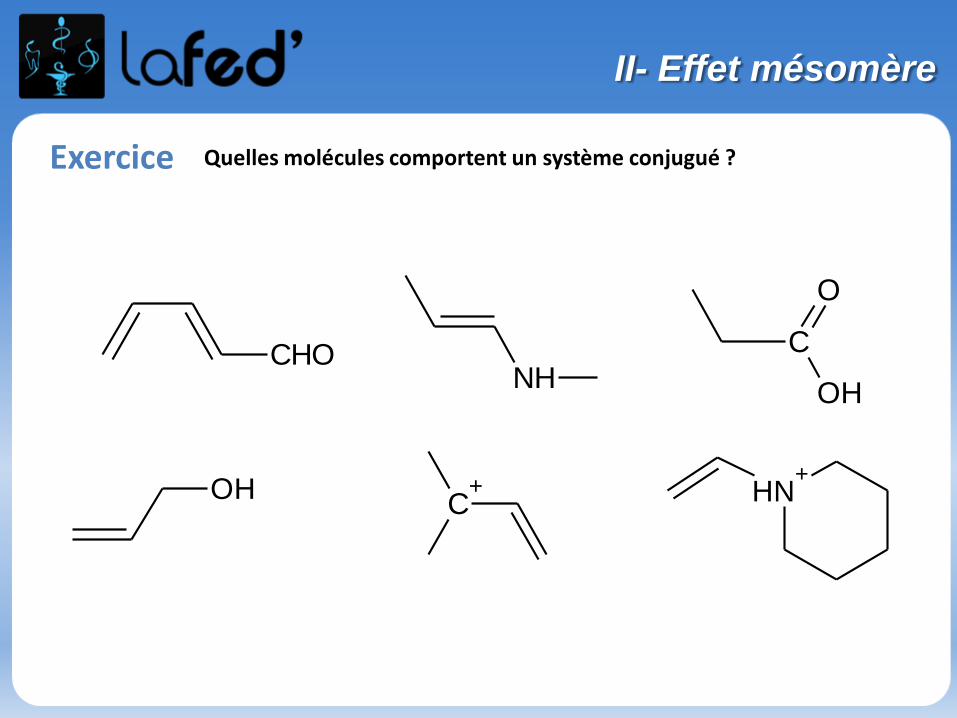
Exercice Quelles molécules comportent un système conjugué ?
NH
OH
CHO
NH+
C
OH
O
C+
II- Effet mésomère
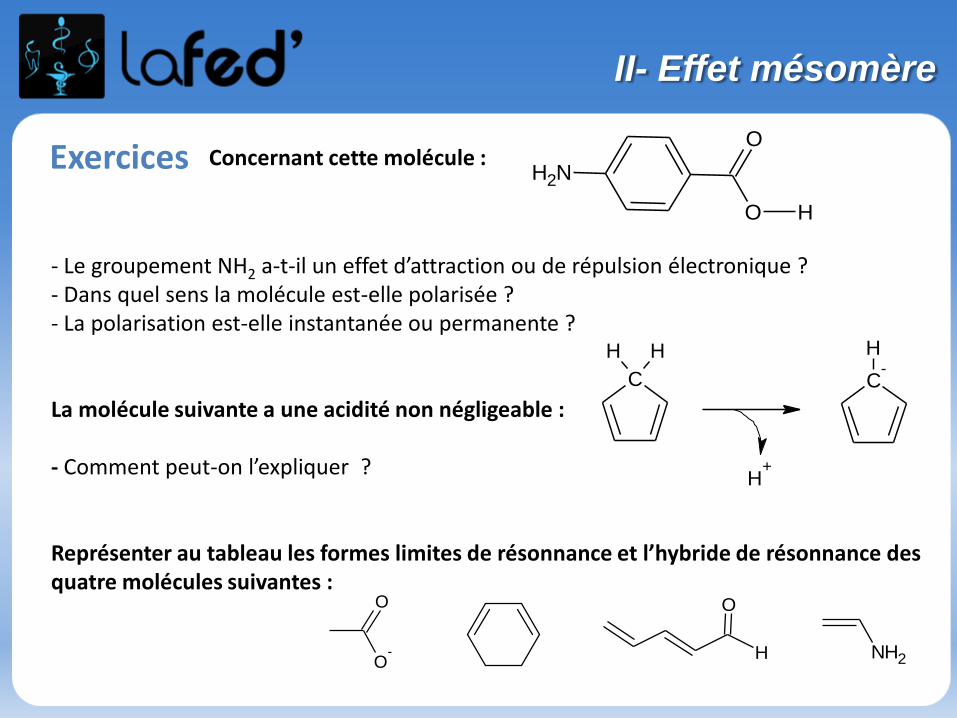
Exercices Concernant cette molécule :NH2
O
O
H
- Le groupement NH2 a-t-il un effet d’attraction ou de répulsion électronique ? - Dans quel sens la molécule est-elle polarisée ?- La polarisation est-elle instantanée ou permanente ?
La molécule suivante a une acidité non négligeable :
- Comment peut-on l’expliquer ?
Représenter au tableau les formes limites de résonnance et l’hybride de résonnance des quatre molécules suivantes :
C-
H
C
HH
H+
O
HO
-
O
NH2
II- Effet mésomère

Stage de Pré-Rentrée 2012UE 1 – Mécanismes réactionnels
122
Diaporama réalisé par les tuteurs de La Fed’

Lors d’une réaction chimique, un composé initial est attaqué par un réactif pourformer un produit final.
Il y a rupture de liaisons (ex : rupture d’une double liaison π qui devient simpleliaison σ après une addition électrophile) et/ou formation de nouvelles liaisons.
Il peut y avoir une ou plusieurs étapes.
Il existe des mécanismes fondamentaux (addition, élimination, substitution) quipeuvent se succéder lors de réactions plus complexes (ex : estérification =addition nucléophile suivie d’une élimination).
I- Introduction

I-Introduction
II-Principaux mécanismes réactionnels
1-Addition2-Substitution3-Elimination
II-Exemples de réaction
Pré-requis indispensables à la compréhension du cours.Définitions et bases à maitriser +++
Estérification, amidification, acétalisation
I- Introduction

Notion de fonction chimiqueLes molécules (très diverses) peuvent être regroupées en fonction de leursréactivité chimique. Cette réactivité dépend de fonctions bien particulières àconnaître. Elles sont explicitées sur une formule topologique (-OH, -COOH…)
Un squelette carboné saturé ne présente pas de réactivité chimique particulière.
Par exemple : présentera la même réactivité que :
et on pourra réaliser en théorie les mêmes réactions chimiques.
Cependant, les propriétés physiques peuvent différer (solubilité, température d’ébullition) ce qui peut rendre les réactions plus ou moins compliquées.
OHOH
I- a) Fonctions chimiques
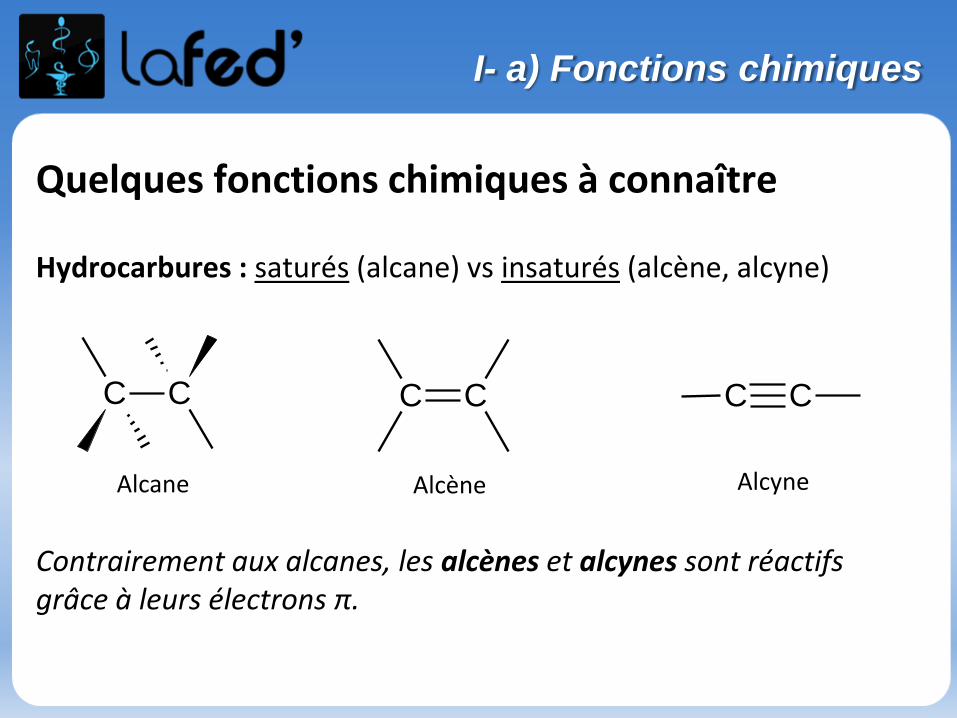
Quelques fonctions chimiques à connaître
Hydrocarbures : saturés (alcane) vs insaturés (alcène, alcyne)
Contrairement aux alcanes, les alcènes et alcynes sont réactifs grâce à leurs électrons π.
C CC C C C
Alcane Alcène Alcyne
I- a) Fonctions chimiques
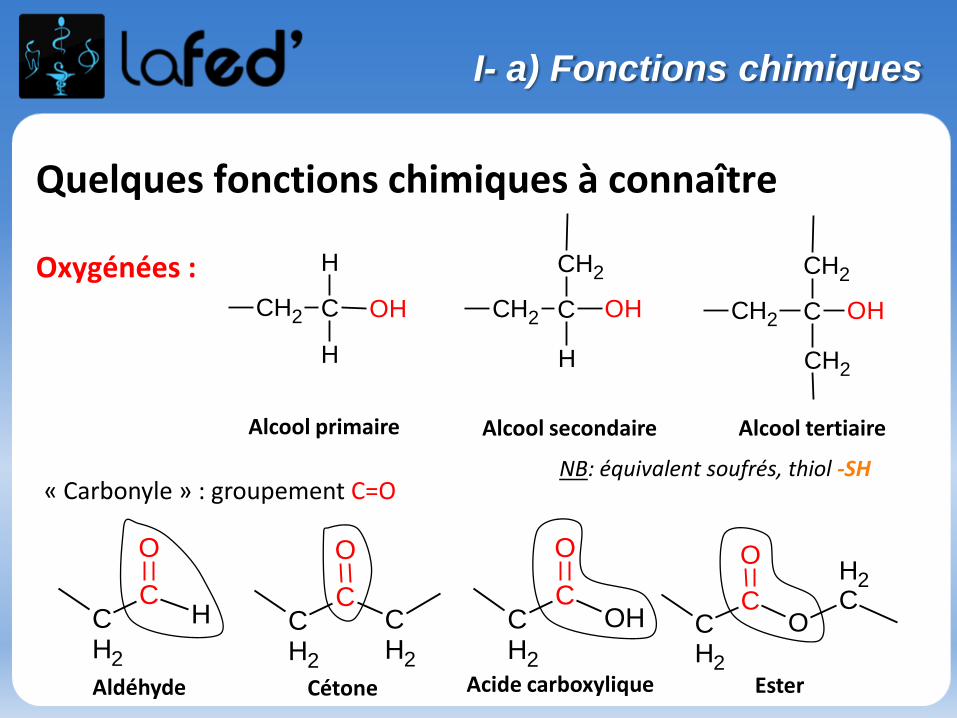
Quelques fonctions chimiques à connaître
Oxygénées : CH2 C OH
CH2
H
CH2 C
H
H
OH CH2 C OH
CH2
CH2
Alcool primaire Alcool secondaire Alcool tertiaire
« Carbonyle » : groupement C=O
Aldéhyde Cétone
CH2
CCH2
O
CH2
COH
O
CH2
CO
O
CH2
CH2
CH
O
Acide carboxylique Ester
NB: équivalent soufrés, thiol -SH
I- a) Fonctions chimiques
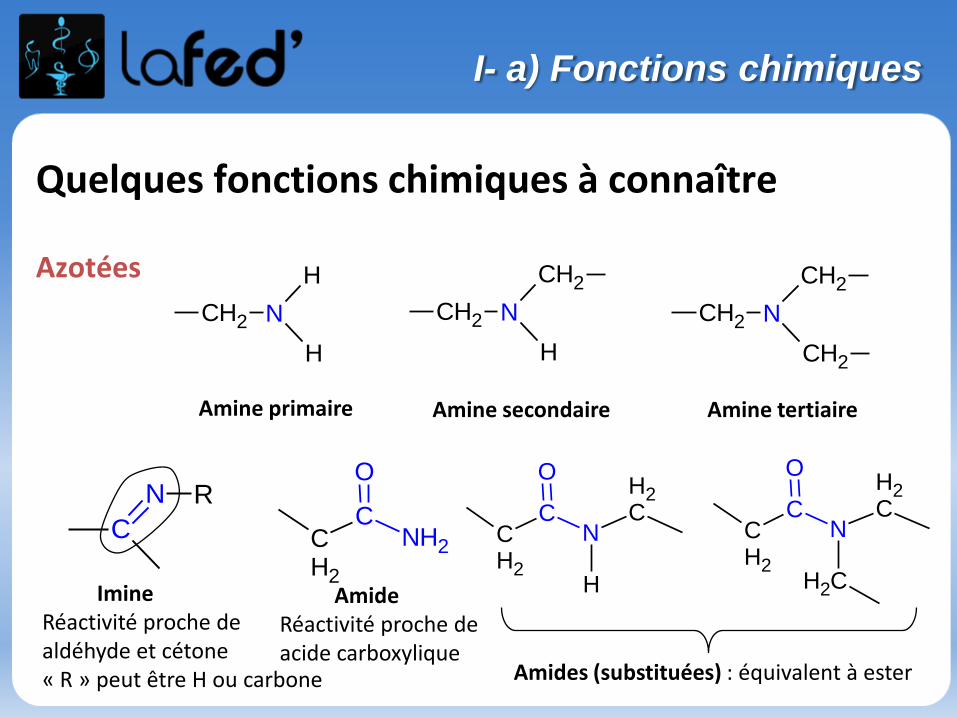
Quelques fonctions chimiques à connaître
Azotées
ImineRéactivité proche de aldéhyde et cétone« R » peut être H ou carbone
CH2 N
H
H
CH2 N
CH2
H
CH2 N
CH2
CH2
Amine primaire Amine secondaire Amine tertiaire
C
N R
CH2
CN
O
CH2
H
CH2
CN
O
CH2
CH2
CH2
CNH2
O
AmideRéactivité proche de acide carboxylique
Amides (substituées) : équivalent à ester
I- a) Fonctions chimiques
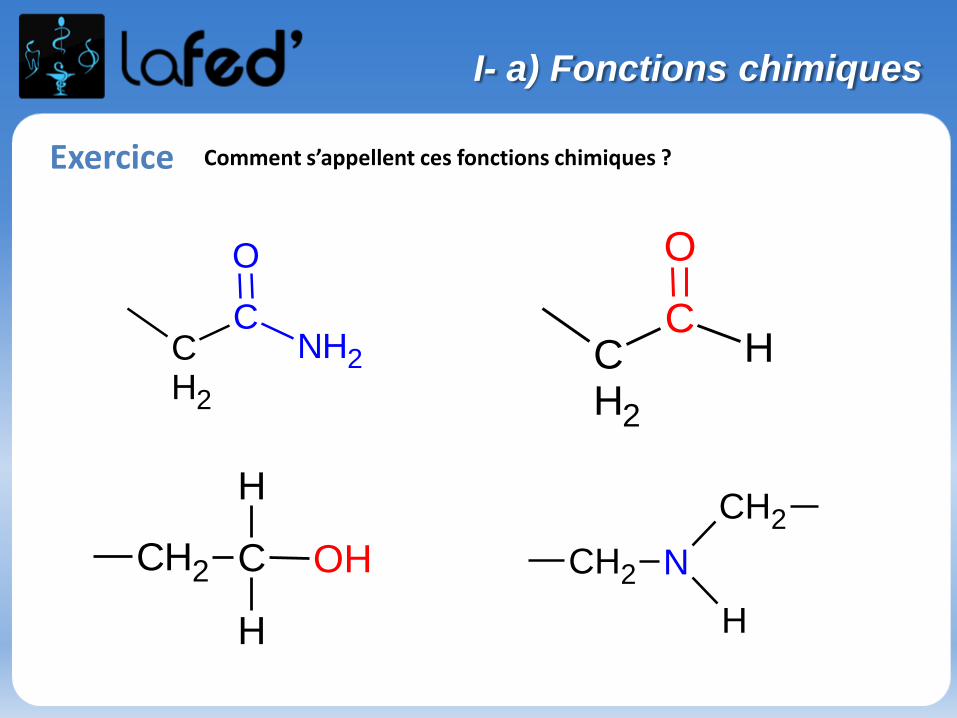
Exercice Comment s’appellent ces fonctions chimiques ?
CH2
CNH2
O
CH2
CH
O
CH2 C
H
H
OH CH2 N
CH2
H
I- a) Fonctions chimiques
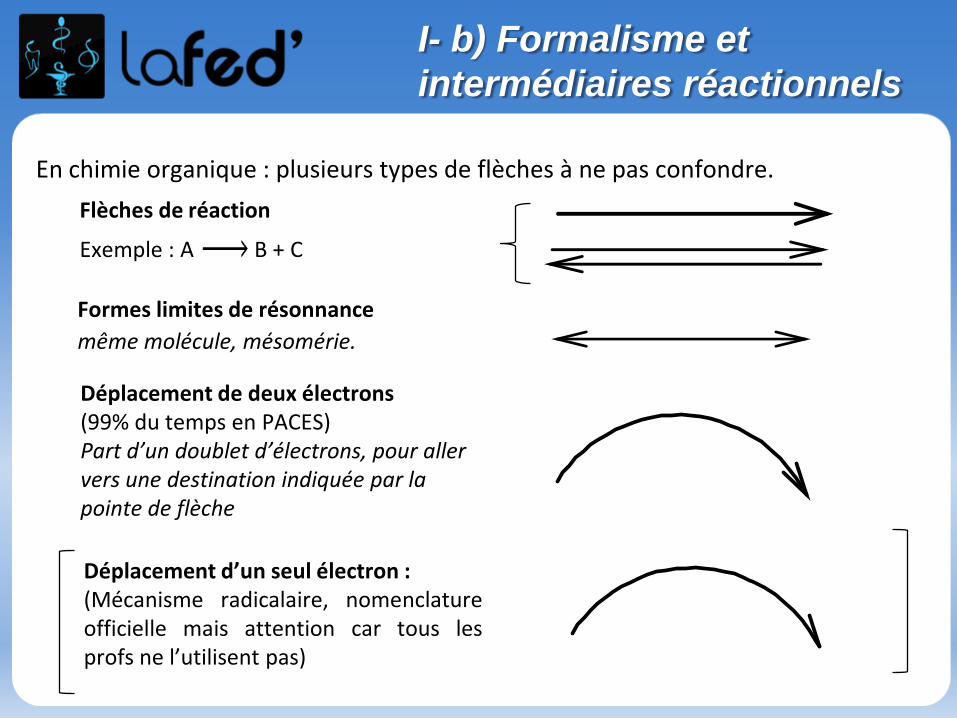
En chimie organique : plusieurs types de flèches à ne pas confondre.
Flèches de réaction
Exemple : A B + C
Formes limites de résonnance
même molécule, mésomérie.
Déplacement de deux électrons (99% du temps en PACES)Part d’un doublet d’électrons, pour aller vers une destination indiquée par la pointe de flèche
Déplacement d’un seul électron : (Mécanisme radicalaire, nomenclatureofficielle mais attention car tous lesprofs ne l’utilisent pas)
I- b) Formalisme et
intermédiaires réactionnels
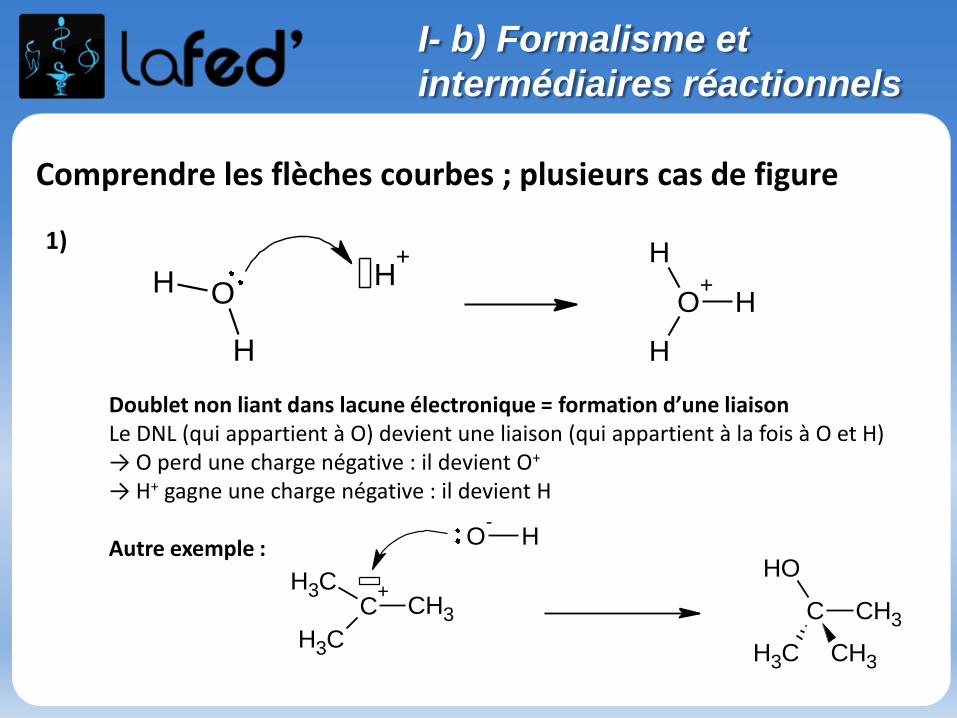
Comprendre les flèches courbes ; plusieurs cas de figure
OH
H
H+
O+
H
H
H
1)
Doublet non liant dans lacune électronique = formation d’une liaisonLe DNL (qui appartient à O) devient une liaison (qui appartient à la fois à O et H)→ O perd une charge négative : il devient O+
→ H+ gagne une charge négative : il devient H
Autre exemple :
C+
CH3
CH3
CH3
O-
H
C CH3
OH
CH3 CH3
I- b) Formalisme et
intermédiaires réactionnels
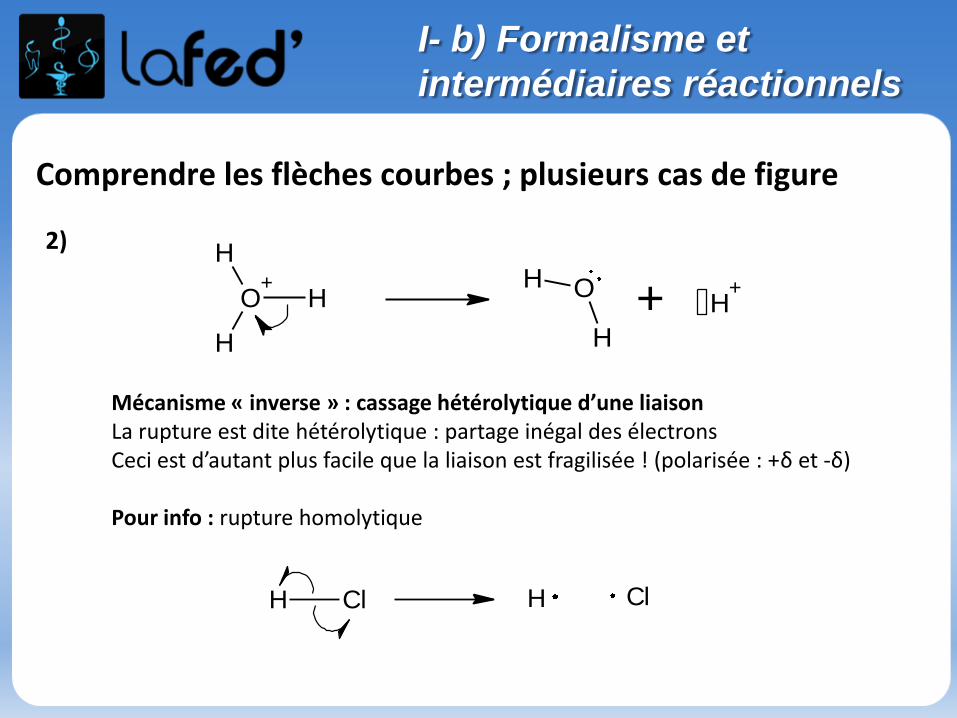
Comprendre les flèches courbes ; plusieurs cas de figure
2)
Mécanisme « inverse » : cassage hétérolytique d’une liaisonLa rupture est dite hétérolytique : partage inégal des électronsCeci est d’autant plus facile que la liaison est fragilisée ! (polarisée : +δ et -δ)
Pour info : rupture homolytique
O+
H
H
H
OH
H
+ H+
H Cl H Cl
I- b) Formalisme et
intermédiaires réactionnels
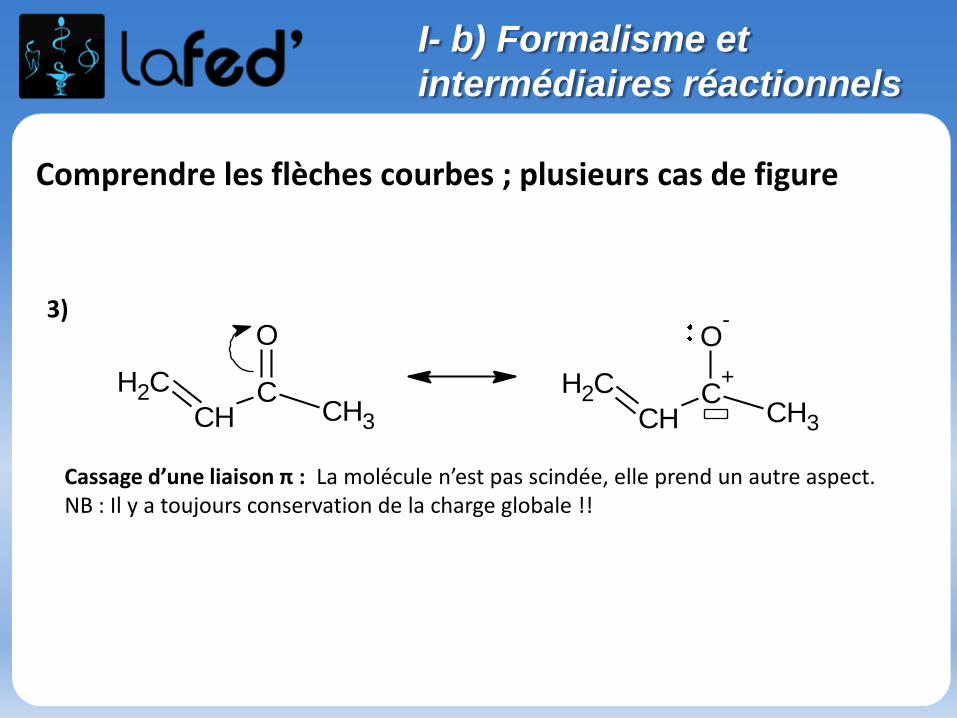
Comprendre les flèches courbes ; plusieurs cas de figure
3)
Cassage d’une liaison π : La molécule n’est pas scindée, elle prend un autre aspect. NB : Il y a toujours conservation de la charge globale !!
CHC
CH3
O
CH2
CHC
+
CH3
O-
CH2
I- b) Formalisme et
intermédiaires réactionnels
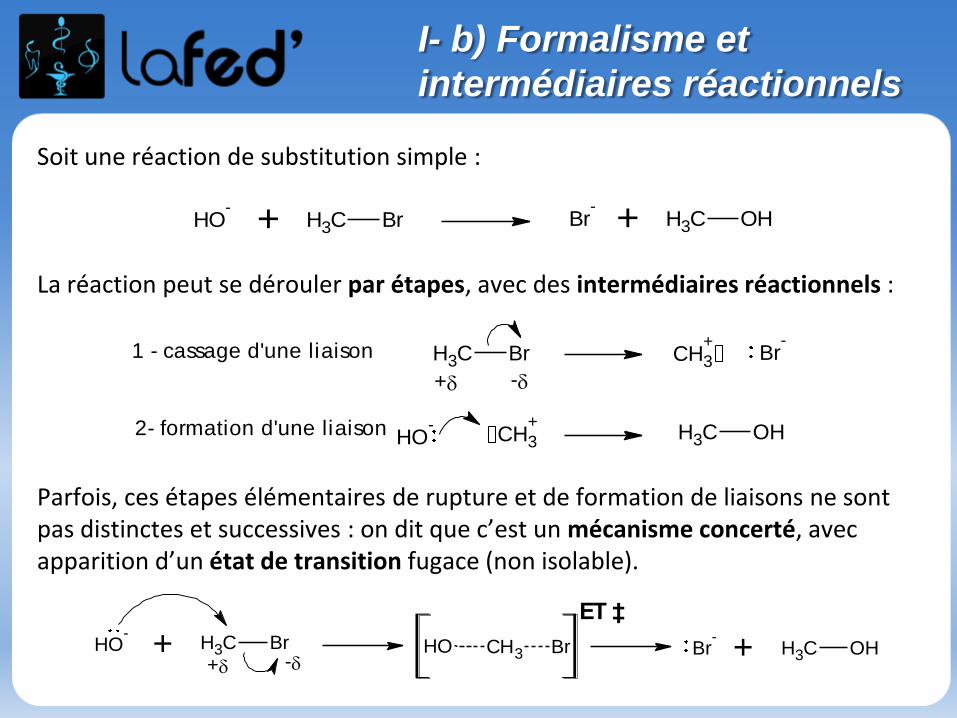
Soit une réaction de substitution simple :
La réaction peut se dérouler par étapes, avec des intermédiaires réactionnels :
Parfois, ces étapes élémentaires de rupture et de formation de liaisons ne sont pas distinctes et successives : on dit que c’est un mécanisme concerté, avec apparition d’un état de transition fugace (non isolable).
CH3 BrOH-
+ CH3 OHBr-
+
CH3 Br
-+
CH3
+Br
-
CH3
+OH
- CH3 OH
1 - cassage d'une liaison
2- formation d'une liaison
CH3 BrOH-
+ CH3 OHBr-
++ -
OH CH3 Br
ET ‡
I- b) Formalisme et
intermédiaires réactionnels
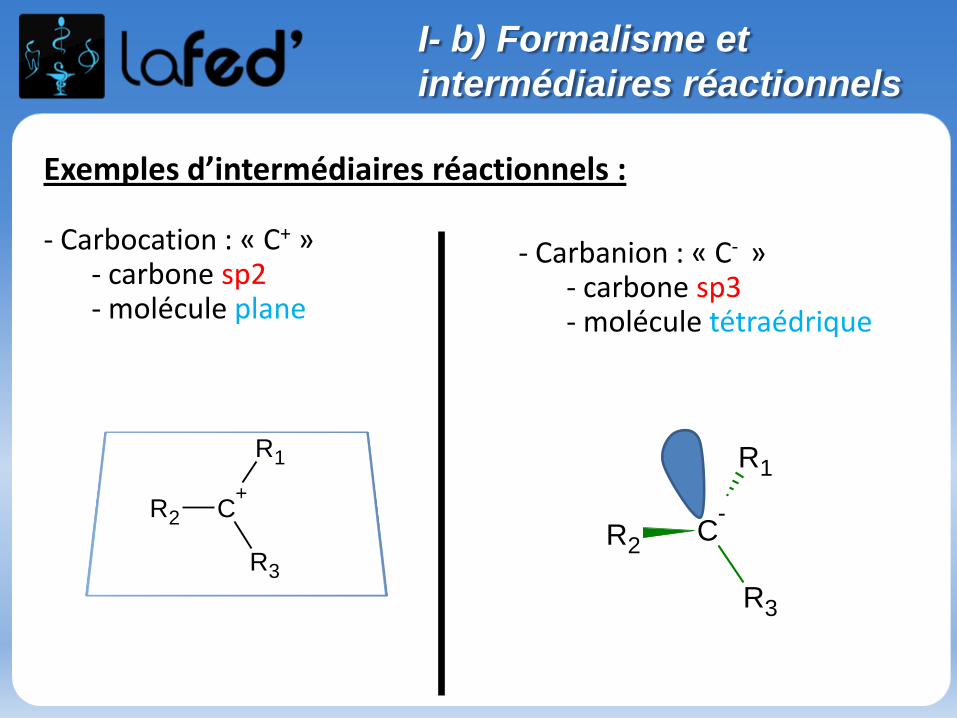
Exemples d’intermédiaires réactionnels :
- Carbocation : « C+ »- carbone sp2- molécule plane
- Carbanion : « C- »- carbone sp3- molécule tétraédrique
C+
R1
R2
R3
C-
R1
R2
R3
I- b) Formalisme et
intermédiaires réactionnels
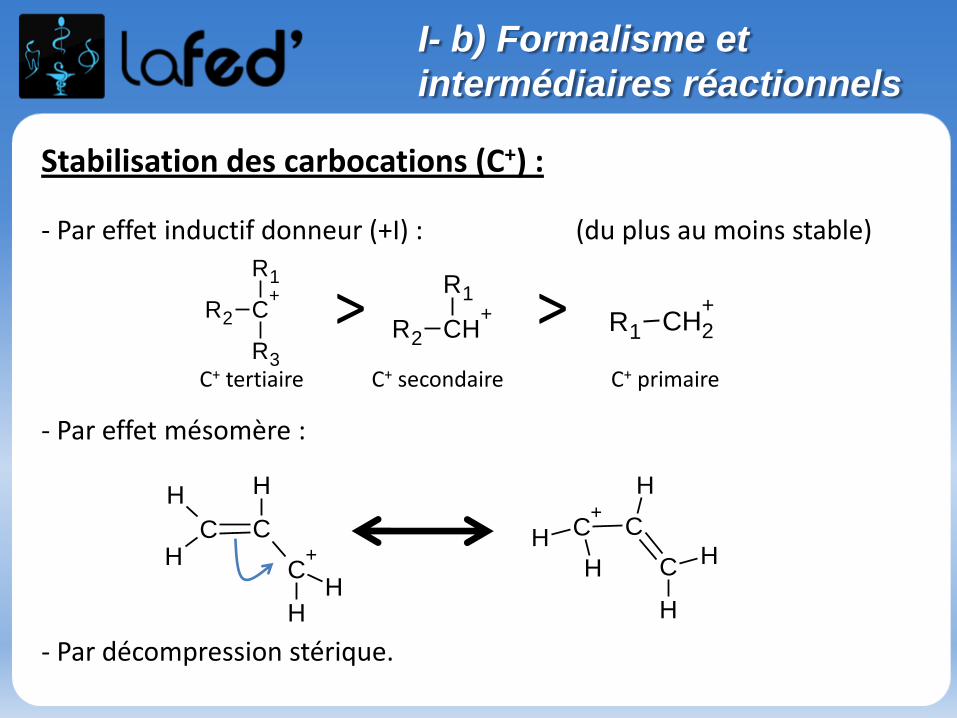
I- b) Formalisme et
intermédiaires réactionnels
Stabilisation des carbocations (C+) :
- Par effet inductif donneur (+I) : (du plus au moins stable)
- Par effet mésomère :
- Par décompression stérique.
C+
R1
R2
R3
CH+
R1
R2CH2
+R1
C C
C+
H
HH
H
H
C+
C
C
H
H
H
H
H
C+ tertiaire C+ secondaire C+ primaire
> >
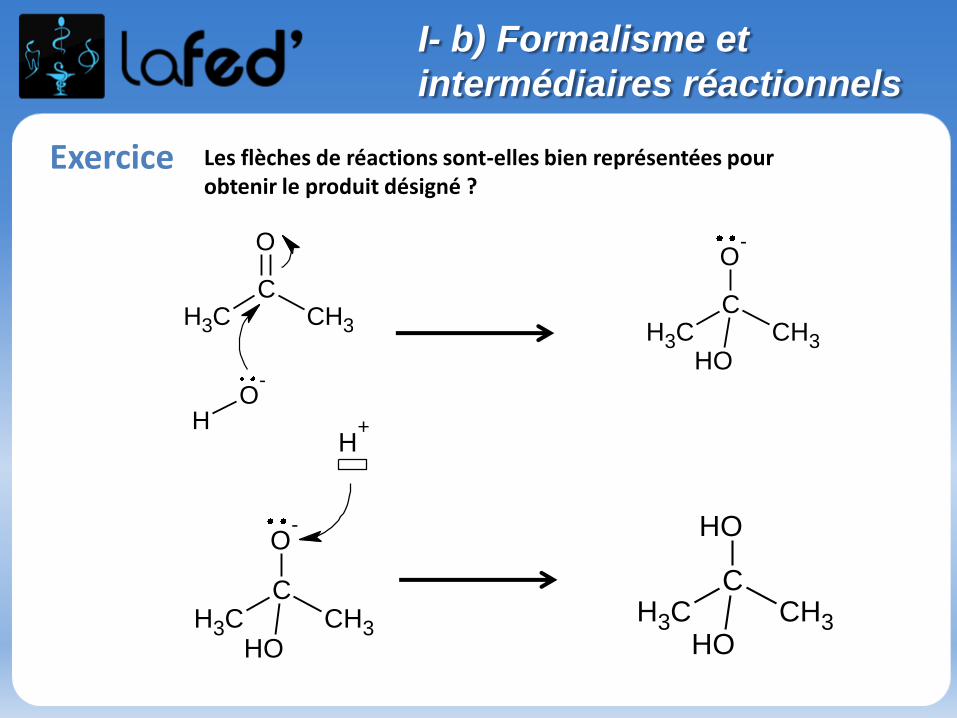
Exercice Les flèches de réactions sont-elles bien représentées pour obtenir le produit désigné ?
CH3
CCH3
O
O-
H
CH3
CCH3
O-
OH
CH3
CCH3
O-
OH
H+
CH3
CCH3
OH
OH
I- b) Formalisme et
intermédiaires réactionnels
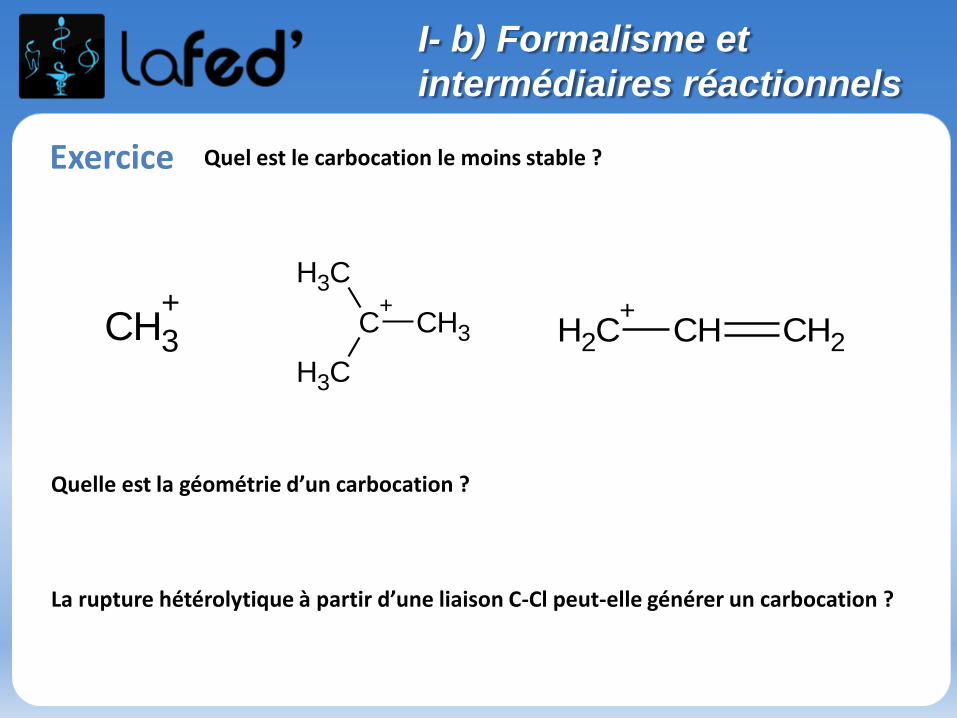
Exercice Quel est le carbocation le moins stable ?
CH3
+C
+CH3
CH3
CH3
CH2
+CH CH2
I- b) Formalisme et
intermédiaires réactionnels
Quelle est la géométrie d’un carbocation ?
La rupture hétérolytique à partir d’une liaison C-Cl peut-elle générer un carbocation ?

Un nucléophile Nu est un composé riche en électrons
Plus précisément, toute espèce possédant un doublet électronique réactif : bases deLewis, anions. Les nucléophiles sont attirés par les charges +
Exemples :
Un électrophile E est un composé pauvre en électrons, attiré par les charges –
Ces espèces possèdent parfois une lacune électronique (acides de Lewis, cations). Ellespossèdent au moins un déficit électronique (+δ).
La plupart du temps, ce sont surtout des « sites électrophiles »
Définitions à comprendre ++++++++++
O
CCH3 CH3
CH3 Cl C+
CH3
CH3
CH3
+ --
+
C-
N O-
H mais aussiO
H
HCH3 NH2
I- c) Définitions
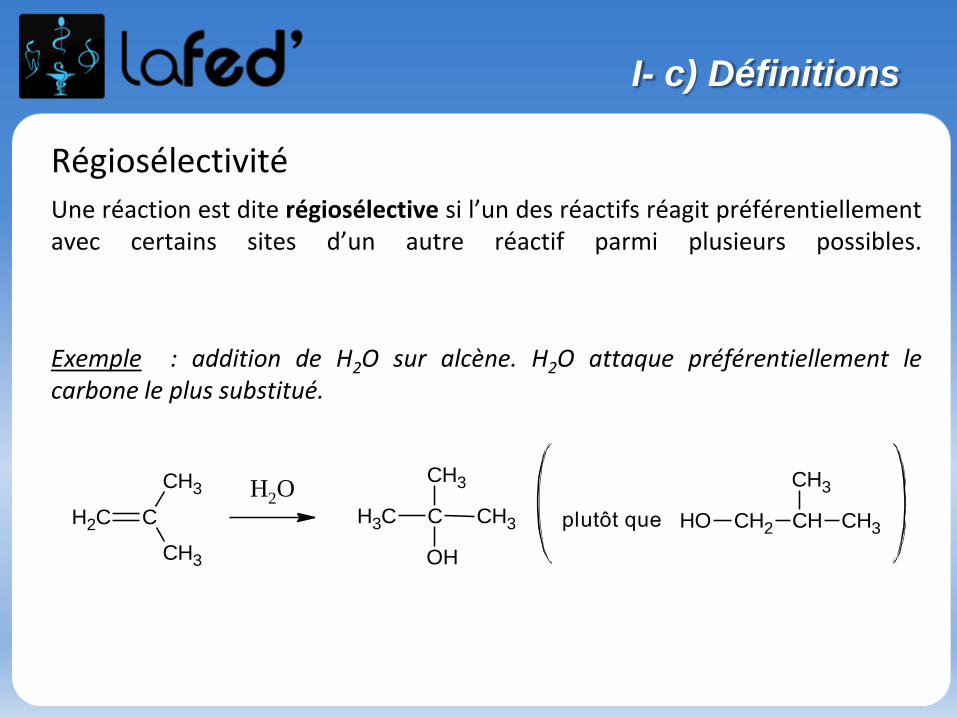
RégiosélectivitéUne réaction est dite régiosélective si l’un des réactifs réagit préférentiellementavec certains sites d’un autre réactif parmi plusieurs possibles.
Exemple : addition de H2O sur alcène. H2O attaque préférentiellement lecarbone le plus substitué.
CH2 C
CH3
CH3
CH3 C CH3
CH3
OH
H2O
plutôt que CH2 CH CH3
CH3
OH
I- c) Définitions
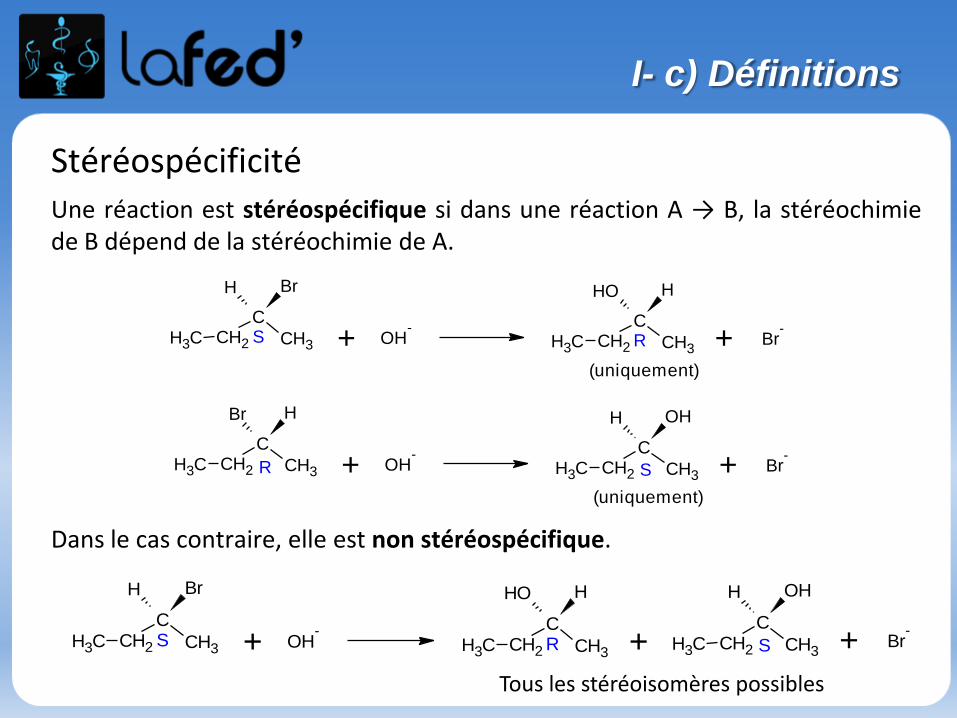
StéréospécificitéUne réaction est stéréospécifique si dans une réaction A → B, la stéréochimiede B dépend de la stéréochimie de A.
Dans le cas contraire, elle est non stéréospécifique.
CH2
C
CH3
BrH
CH3 S OH-
+ CH2
C
CH3
HOH
CH3 R + Br-
(uniquement)
CH2
C
CH3
HBr
CH3 SOH-
+ CH2
C
CH3
OHH
CH3R + Br-
(uniquement)
CH2
C
CH3
BrH
CH3 S OH-
+ CH2
C
CH3
HOH
CH3 R + SCH2
C
CH3
OHH
CH3 + Br-
Tous les stéréoisomères possibles
I- c) Définitions
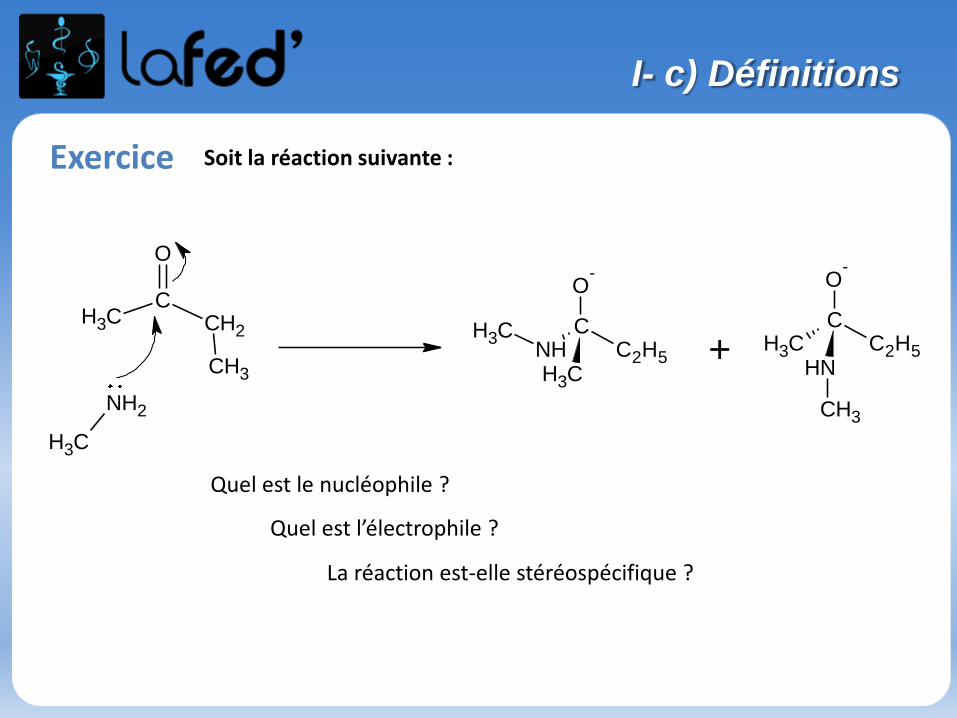
Exercice Soit la réaction suivante :
Quel est le nucléophile ?
Quel est l’électrophile ?
La réaction est-elle stéréospécifique ?
CH3
CCH2
O
CH3
NH2
CH3
NHC
C2H5
O-
CH3
CH3 CH3
CC2H5
O-
NH
CH3
+
I- c) Définitions

143
Additions : A + B → CAu programme de PACES:
Additions nucléophiles et additions électrophiles sur double liaison.
Aujourd’hui, nous ne traiterons pas les additions électrophiles (= Mécanismes les plus compliqués du programme).
Substitutions : A + B → C + DSubstitutions nucléophiles de type 1
Substitutions nucléophiles de type 2
Eliminations : A → B + CEliminations de type 1
Eliminations de type 2
II- Mécanismes réactionnels

144
a) Addition
S’effectue sur une insaturation.
Il y a cassure d’une liaison π.
Il en existe 2 types : Addition électrophile et addition nucléophile
(selon la nature du réactif qui attaque en premier)
II- a) Additions nucléophiles

145
a) Addition
S’effectue sur une insaturation.
Il y a cassure d’une liaison π.
Il en existe 2 types : Addition électrophile et addition nucléophile
(selon la nature du réactif qui attaque en premier)
II- a) Additions nucléophiles

Addition nucléophileS’effectue sur des composés avec insaturation fortement polarisée :
A=A’ où χA’ > χA
Exemples : C=O, C=N-H, C≡N, N=O, etc…
Ce sont des fonctions électrophiles.
Sur A s’additionne un nucléophile, quelques exemples :
H2O: N≡C-: HO-: CH3 O-: H3N: CH3-HN:-CH3 CH3-S:H H-:
Réaction Non Stéréospécifique.
II- a) Additions nucléophiles
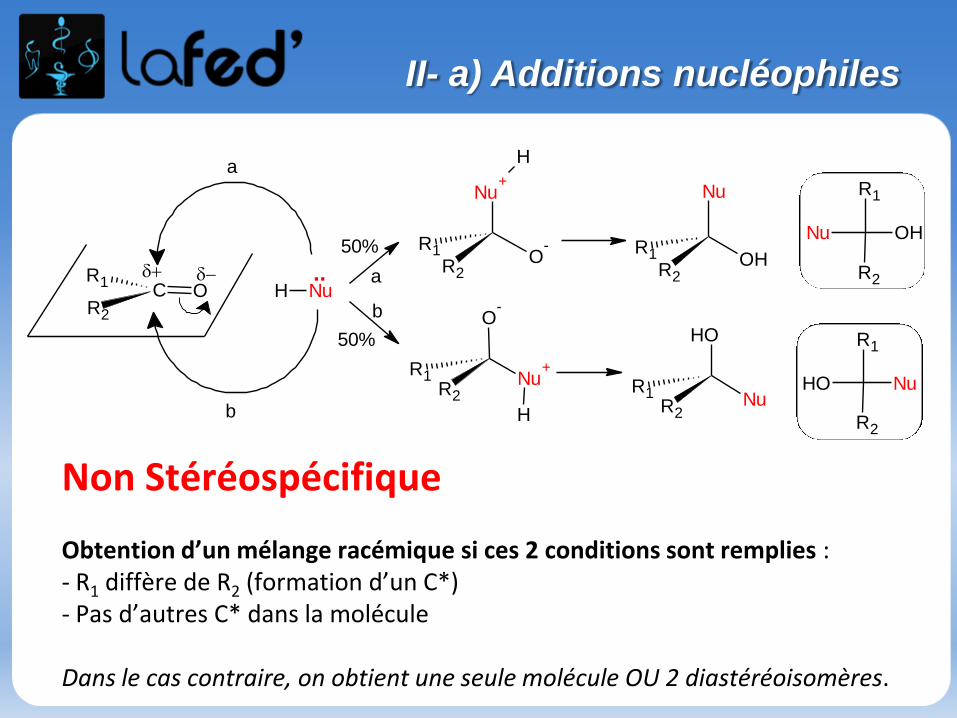
II- a) Additions nucléophiles
Non Stéréospécifique
Obtention d’un mélange racémique si ces 2 conditions sont remplies : - R1 diffère de R2 (formation d’un C*)- Pas d’autres C* dans la molécule
Dans le cas contraire, on obtient une seule molécule OU 2 diastéréoisomères.
R1C O
R2
H Nu
..
a
b
a
b
R1 O-
R2
Nu+
H
R1
O-
R2Nu
+
H
R1 OHR2
Nu
R1
OH
R2Nu
R1
R2
OHNu
R1
R2
NuOH
50%
50%
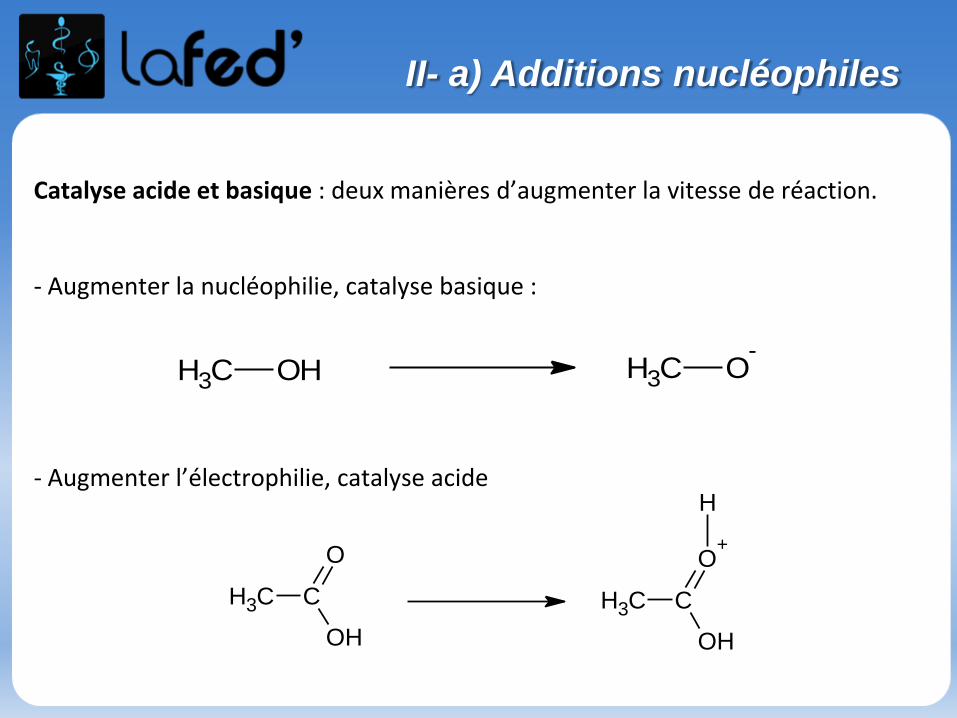
II- a) Additions nucléophiles
Catalyse acide et basique : deux manières d’augmenter la vitesse de réaction.
- Augmenter la nucléophilie, catalyse basique :
- Augmenter l’électrophilie, catalyse acide
CH3 OH CH3 O-
CH3 C
OH
O
CH3 C
OH
O+
H
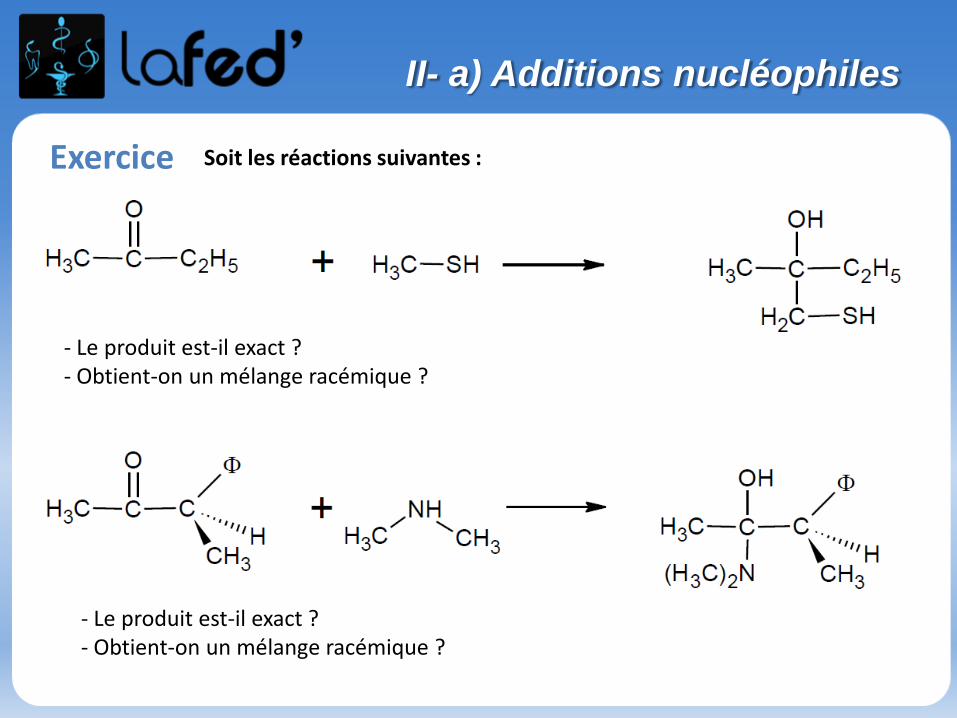
Exercice Soit les réactions suivantes :
- Le produit est-il exact ?- Obtient-on un mélange racémique ?
- Le produit est-il exact ?- Obtient-on un mélange racémique ?
II- a) Additions nucléophiles

b) Substitution
• ll en existe plusieurs type mais nous ne traiterons que la Substitutionnucléophile (SN). (SN = AdN + E)
• Il existe plusieurs mécanismes pour un même bilan.
• La substitution nucléophile s’effectue sur des carbones électrophiles porteurs de nucléofuges (ex : les dérivés halogénés…).
• Il y a remplacement d’un atome (ou d’un groupe d’atomes) par un autre.
II- b) Substitutions nucléophiles
C
R1
R3
R2 X + Nu C
R1
R3
R2 Nu + X- -.. ..
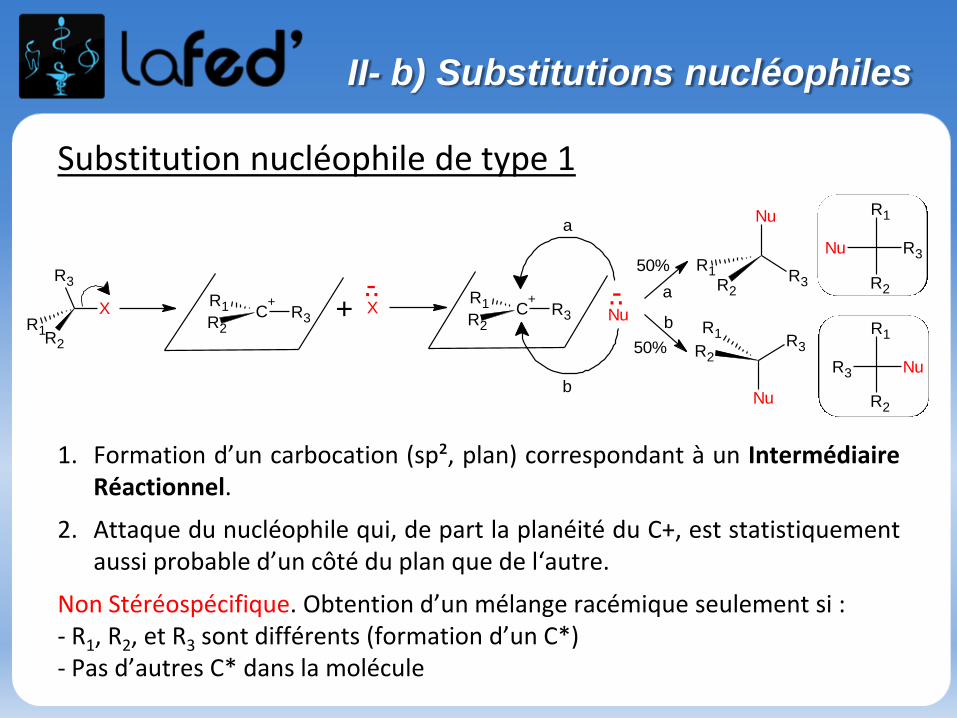
Substitution nucléophile de type 1
1. Formation d’un carbocation (sp², plan) correspondant à un IntermédiaireRéactionnel.
2. Attaque du nucléophile qui, de part la planéité du C+, est statistiquementaussi probable d’un côté du plan que de l‘autre.
Non Stéréospécifique. Obtention d’un mélange racémique seulement si :- R1, R2, et R3 sont différents (formation d’un C*)- Pas d’autres C* dans la molécule
II- b) Substitutions nucléophiles
R1
X
R2
R3
R1 C+
R3R2+ X
..- R1 C+
R3R2Nu-..
a
b
a
b
50%
50%
R1 R3R2
Nu
R1R3R2
Nu
R1
R2
R3Nu
R1
R2
NuR3

SN1 :
2 étapes réactionnelles
Réaction monomoléculaire : v = k[RX]
Favorisée par :
Bonne stabilité du C+ (RX tertiaire, RX secondaireencombré…)
Solvants protiques polaires
Nucléophile faible
Bon nucléofuge
II- b) Substitutions nucléophiles
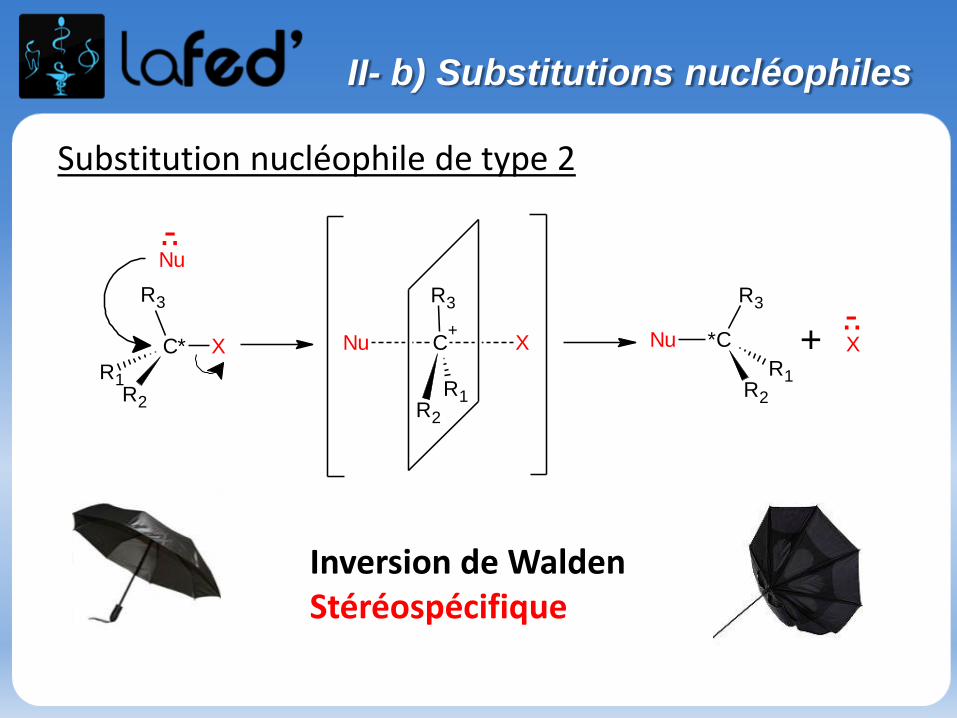
Substitution nucléophile de type 2
II- b) Substitutions nucléophiles
Inversion de WaldenStéréospécifique
..
R1
C* X
R2
R3
Nu-
R1
C+
X
R2
R3
Nu
R1
*CNu
R2
R3
+ X..-

SN2 :
1 seule étape réactionnelle
Réaction bimoléculaire : v = k[Nu][RX]
Favorisée par
C+ peu stable (RX primaire, RX secondaire peu encombré…)
Solvants aprotiques polaires
Nucléophile fort
Bon nucléofuge
II- b) Substitutions nucléophiles
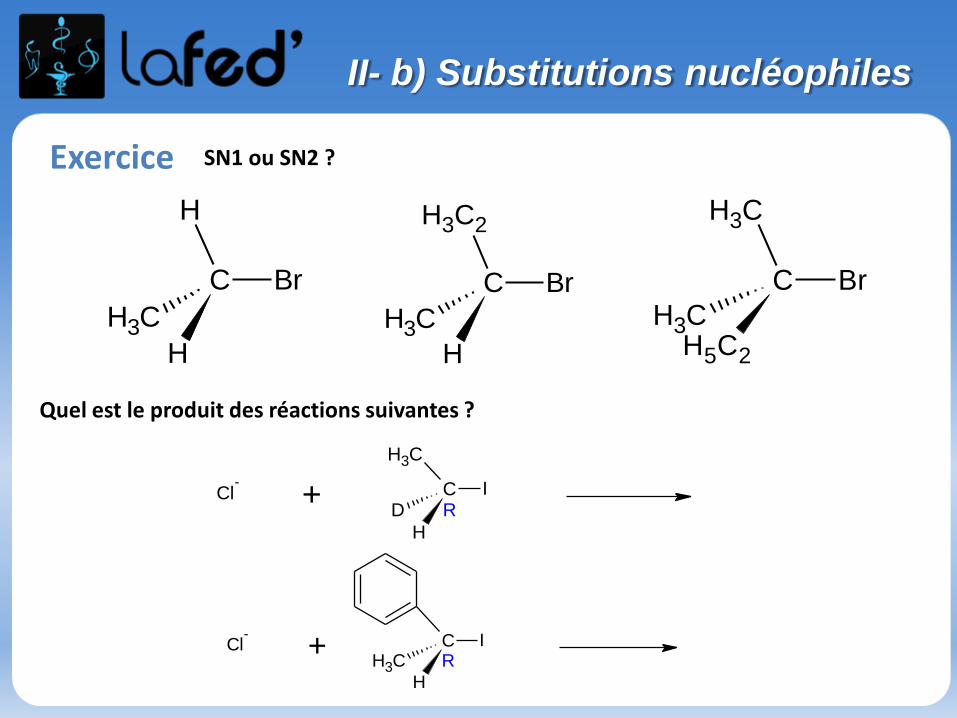
CH3
C Br
H5C2
CH3
CH3
C Br
H
H
CH3
C Br
H
H3C2
Exercice SN1 ou SN2 ?
H
C I
CH3
DCl
-
+R
Quel est le produit des réactions suivantes ?
H
C ICH3
Cl-
+R
II- b) Substitutions nucléophiles

c) Elimination
Il existe 2 types d’Elimination :
- l’Elimination de type 1 (E1)
- l’Elimination de type 2 (E2).
Il y a formation d’une liaison π.
(ex : passage d’un composé saturé à un composé insaturé).
Bilan général : CH3 CH2 OH CH2 CH2 + OH2
II- c) Eliminations
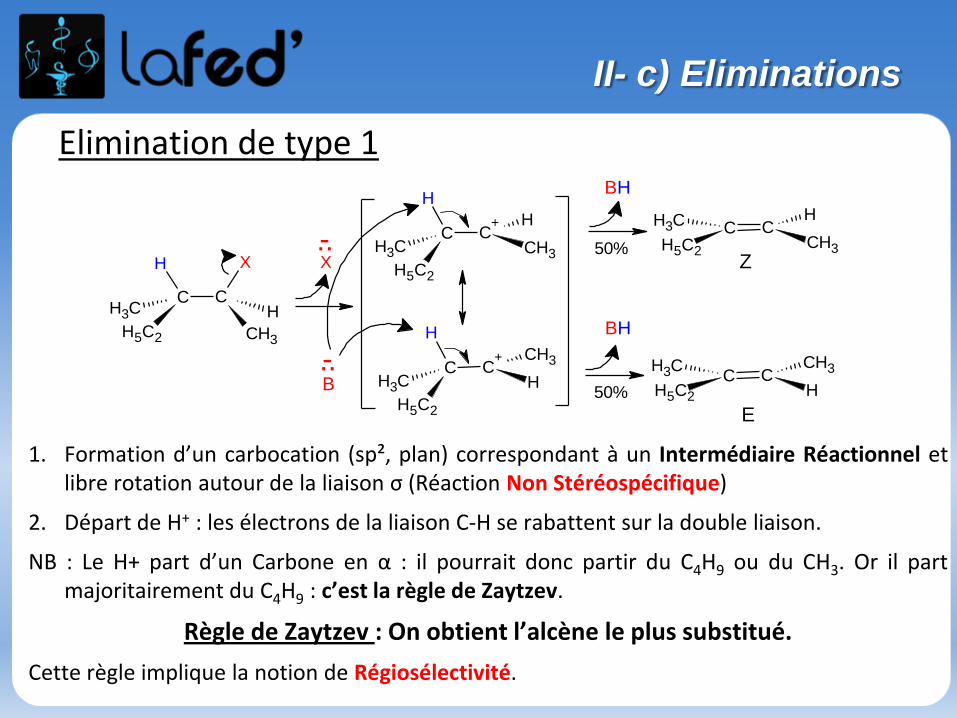
II- c) Eliminations
C CCH3
H5C2
H
CH3
XH X..- C C
+
CH3
H5C2
H
CH3
H
C C+
CH3
H5C2
CH3
H
H
50%
50%
C CCH3
H5C2
H
CH3
C CCH3
H5C2
CH3
H
..-B
BH
BH
Z
E
Elimination de type 1
1. Formation d’un carbocation (sp², plan) correspondant à un Intermédiaire Réactionnel etlibre rotation autour de la liaison σ (Réaction Non Stéréospécifique)
2. Départ de H+ : les électrons de la liaison C-H se rabattent sur la double liaison.
NB : Le H+ part d’un Carbone en α : il pourrait donc partir du C4H9 ou du CH3. Or il partmajoritairement du C4H9 : c’est la règle de Zaytzev.
Règle de Zaytzev : On obtient l’alcène le plus substitué.
Cette règle implique la notion de Régiosélectivité.

158
E1 :
2 étapes réactionnelles
Réaction monomoléculaire : v = k[RX]
Favorisée par :
C+ stable (RX tertiaire, RX secondaire encombré…)
Solvants polaires protiques
Base faible
Bon nucléofuge
On a Régiosélectivité (Zaytzev), mais PAS stéréospécificité (on a Z ET E).
II- c) Eliminations
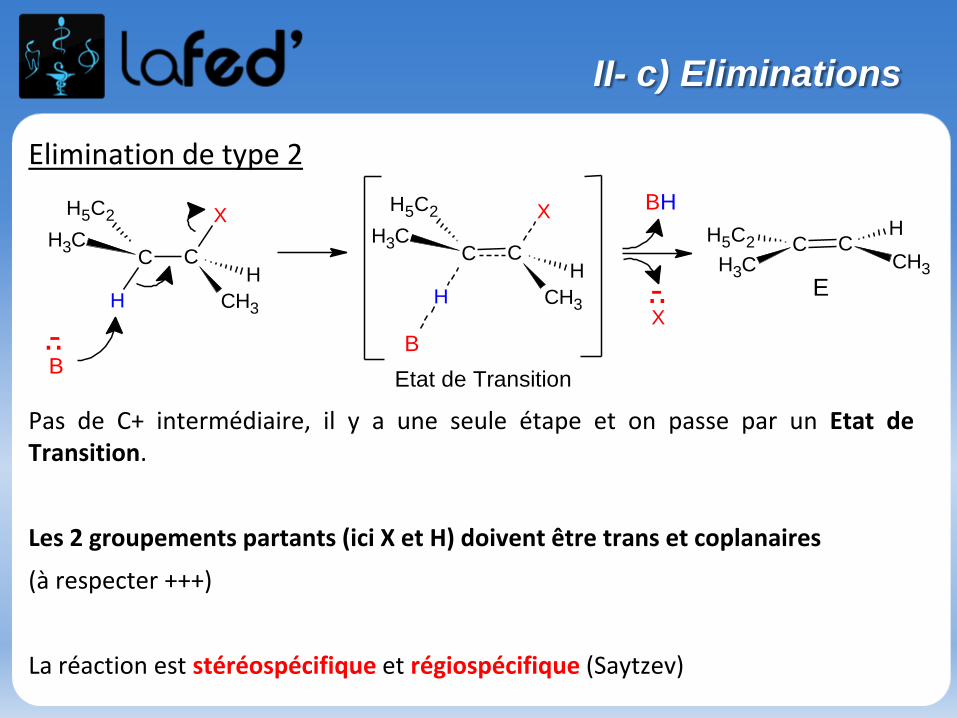
..
..
C C
H5C2
CH3
H
CH3
X
H
-B
C C
H5C2
CH3
H
CH3
X
H
B
Etat de Transition
C CH5C2
CH3
H
CH3
BH
X- E
Elimination de type 2
Pas de C+ intermédiaire, il y a une seule étape et on passe par un Etat deTransition.
Les 2 groupements partants (ici X et H) doivent être trans et coplanaires
(à respecter +++)
La réaction est stéréospécifique et régiospécifique (Saytzev)
II- c) Eliminations

E2 :
1 seule étape réactionnelle
Réaction bimoléculaire : v = k[Nu][RX]
Favorisée par :
Encombrement stérique (RX tertiaire, RX secondaire…) quand compétitionavec SN2
C+ peu stable par rapport à une E1
Solvants aprotiques polaires
Base forte
Bon nucléofuge
On a Régioselectivité ET Stéréospécificité (on a Z OU E).
II- c) Eliminations
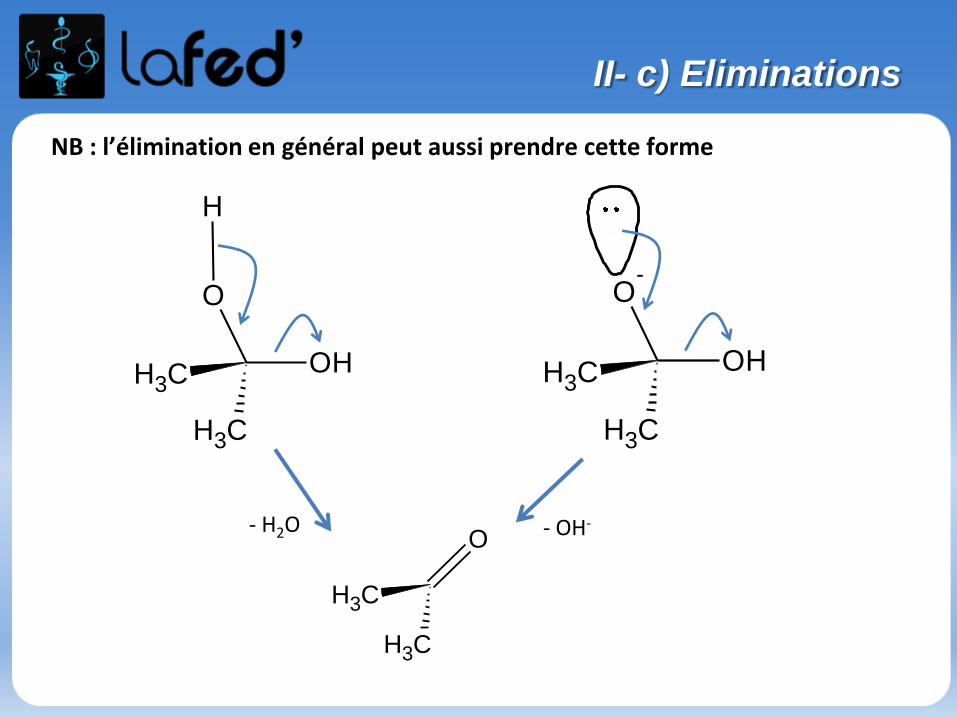
NB : l’élimination en général peut aussi prendre cette forme
II- c) Eliminations
OH
O
CH3
CH3
H
OH
O-
CH3
CH3
- H2O - OH-O
CH3
CH3
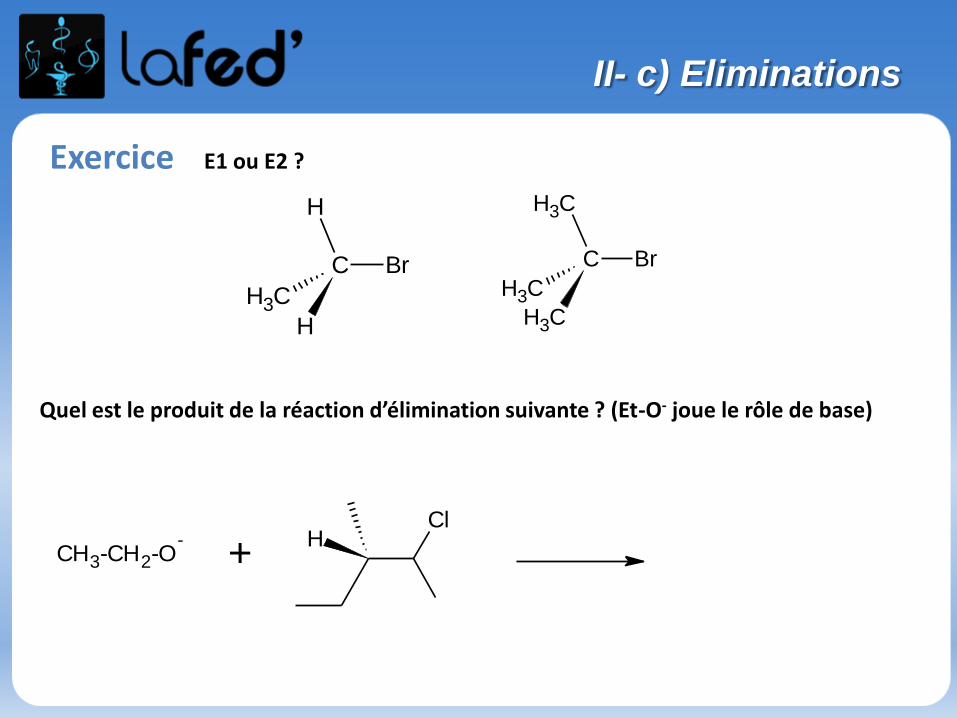
Exercice E1 ou E2 ?
Quel est le produit de la réaction d’élimination suivante ? (Et-O- joue le rôle de base)
CH3
C Br
CH3
CH3
CH3
C Br
H
H
CH3-CH2-O-
+Cl
H
II- c) Eliminations
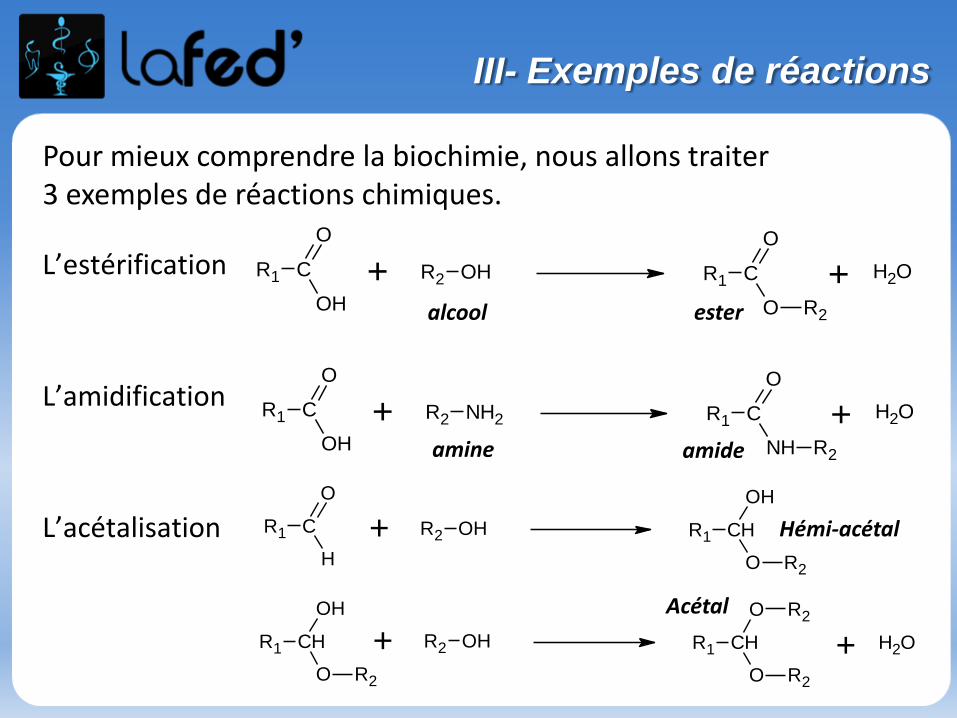
L’estérification
L’amidification
L’acétalisation
Pour mieux comprendre la biochimie, nous allons traiter 3 exemples de réactions chimiques.
R1 C
NH
O
R2
+ OH2R1 C
OH
O
+ R2 NH2
R1 C
O
O
R2
+ OH2R1 C
OH
O
+ R2 OH
alcool
amine
ester
amide
R1 CH
O
OH
R2
R1 C
H
O
+ R2 OH
R1 CH
O
O
R2
R2
+ OH2+ R2 OHR1 CH
O
OH
R2
Hémi-acétal
Acétal
III- Exemples de réactions
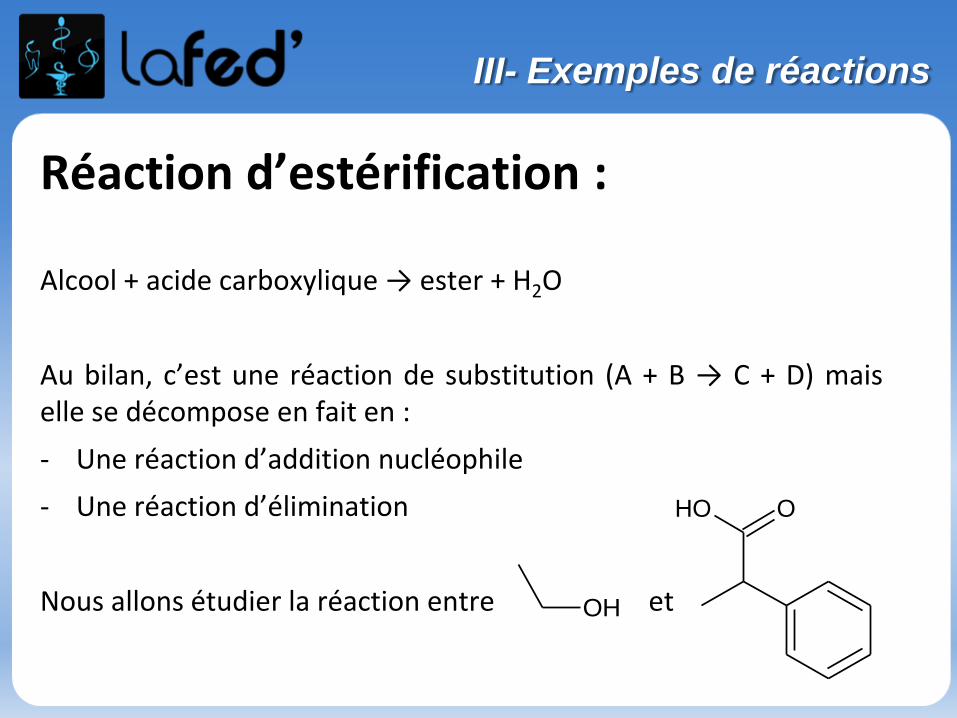
Réaction d’estérification :
Alcool + acide carboxylique → ester + H2O
Au bilan, c’est une réaction de substitution (A + B → C + D) maiselle se décompose en fait en :
- Une réaction d’addition nucléophile
- Une réaction d’élimination
Nous allons étudier la réaction entre et
OOH
OH
III- Exemples de réactions
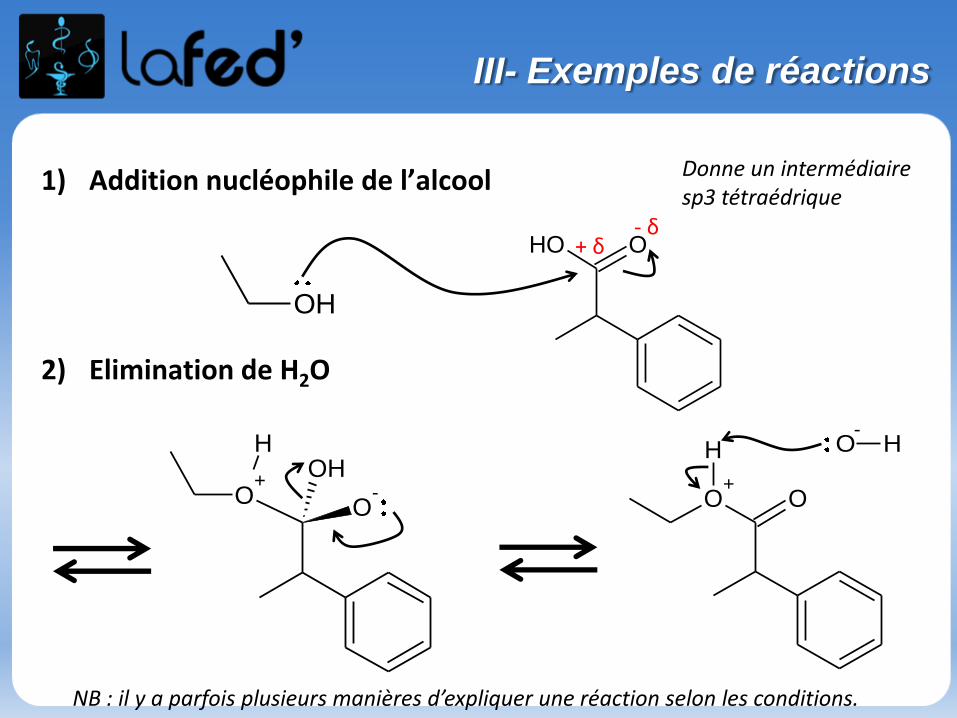
1) Addition nucléophile de l’alcool
2) Elimination de H2O
OH
OOH- δ
+ δ
O+
O-
OHH
O+
O
H O-
H
NB : il y a parfois plusieurs manières d’expliquer une réaction selon les conditions.
Donne un intermédiaire sp3 tétraédrique
III- Exemples de réactions
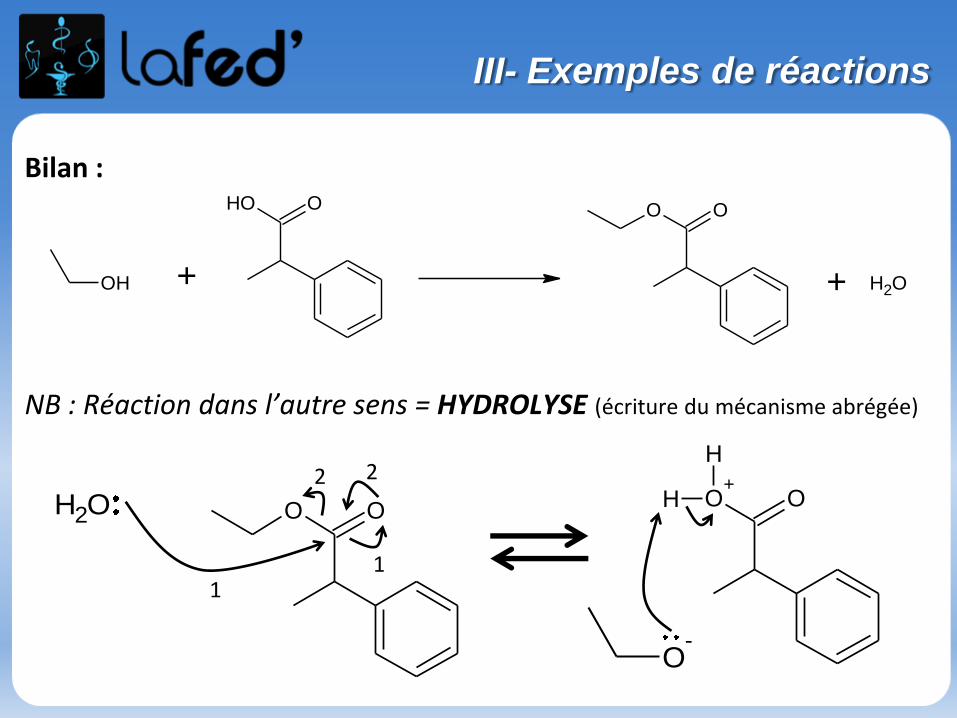
Bilan :
NB : Réaction dans l’autre sens = HYDROLYSE (écriture du mécanisme abrégée)
O O
OH2+OH
OOH
+
OH2 O OO
+OH
H
O-
11
22
III- Exemples de réactions
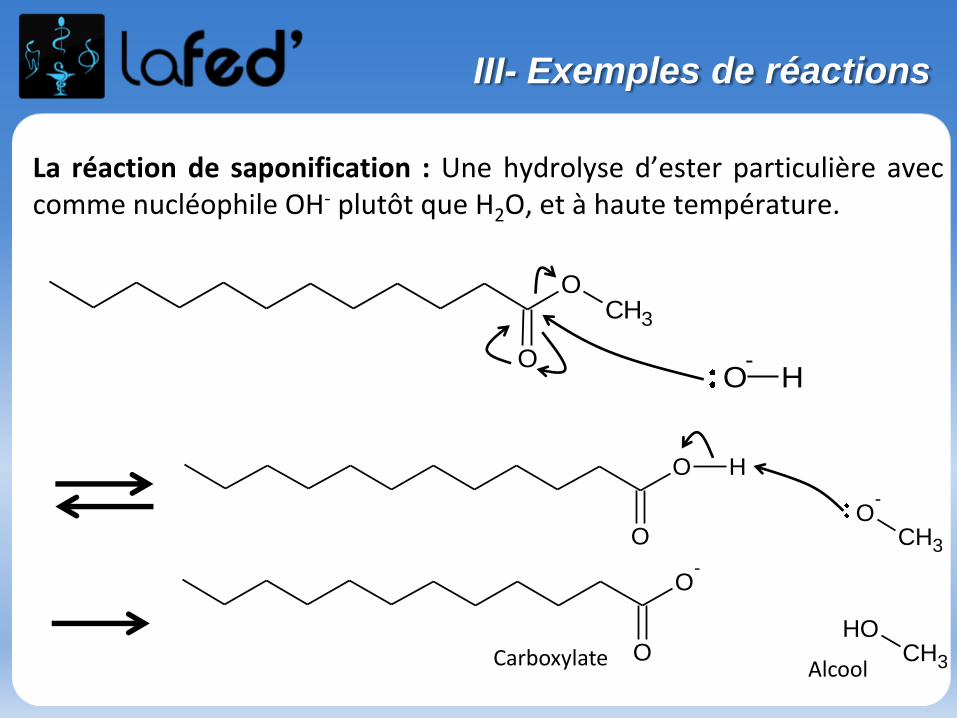
La réaction de saponification : Une hydrolyse d’ester particulière aveccomme nucléophile OH- plutôt que H2O, et à haute température.
O
OCH3
O-
H
O
O H
O-
CH3
O
O-
OHCH3Carboxylate Alcool
III- Exemples de réactions
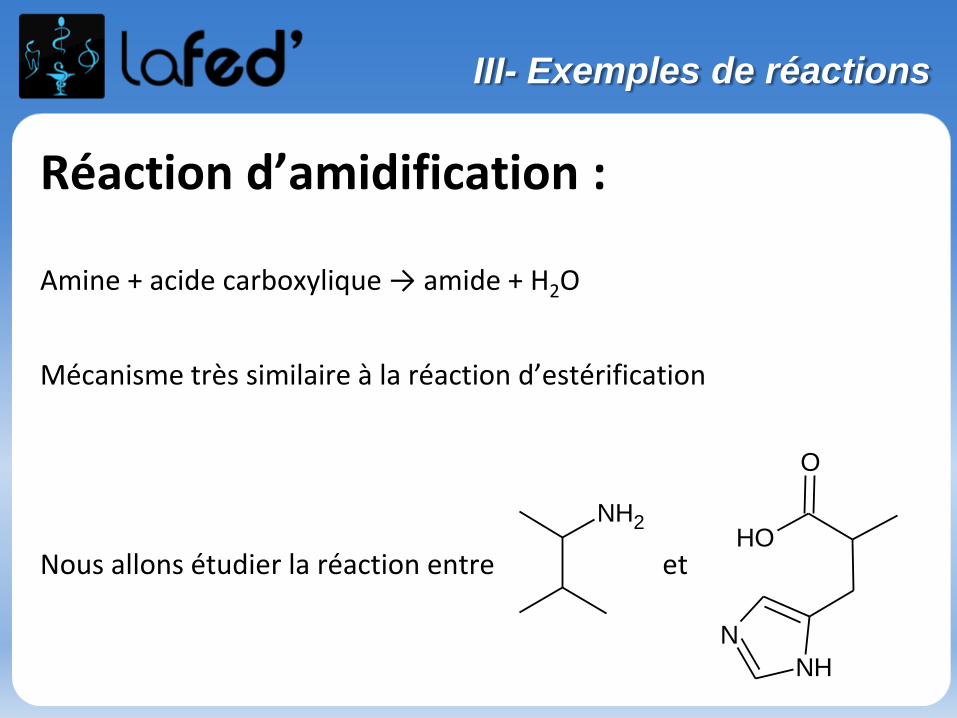
Réaction d’amidification :
Amine + acide carboxylique → amide + H2O
Mécanisme très similaire à la réaction d’estérification
Nous allons étudier la réaction entre et
NH2
O
OH
NH
N
III- Exemples de réactions
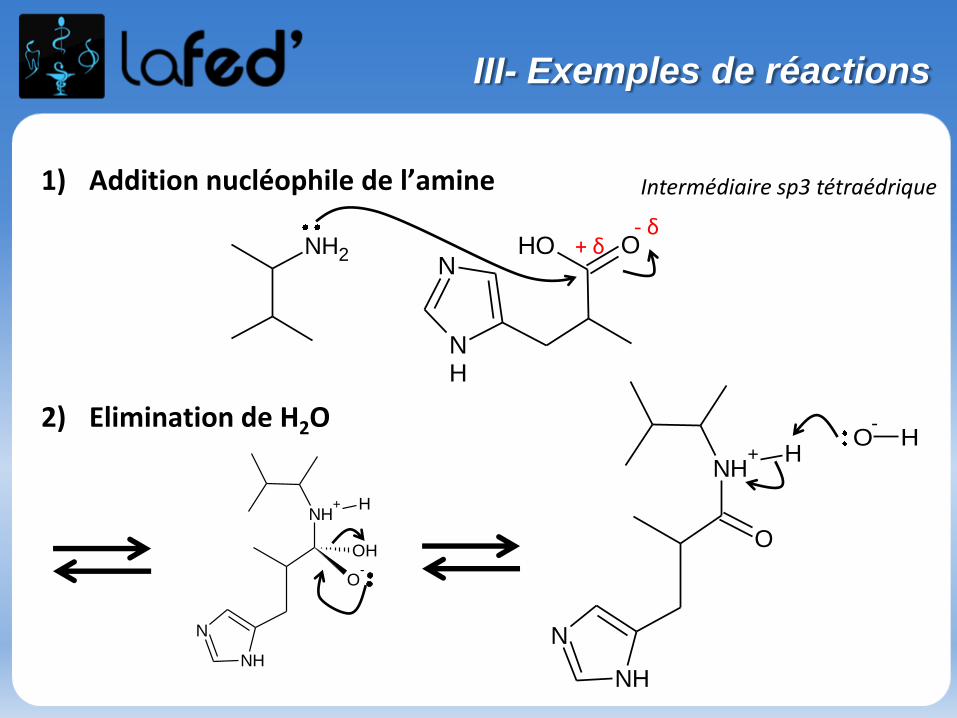
1) Addition nucléophile de l’amine
2) Elimination de H2O
- δ+ δ
O-
H
Intermédiaire sp3 tétraédrique
NH2 OOH
NH
N
NH+
O-
OH
NH
N
H
NH+
NH
N
H
O
III- Exemples de réactions
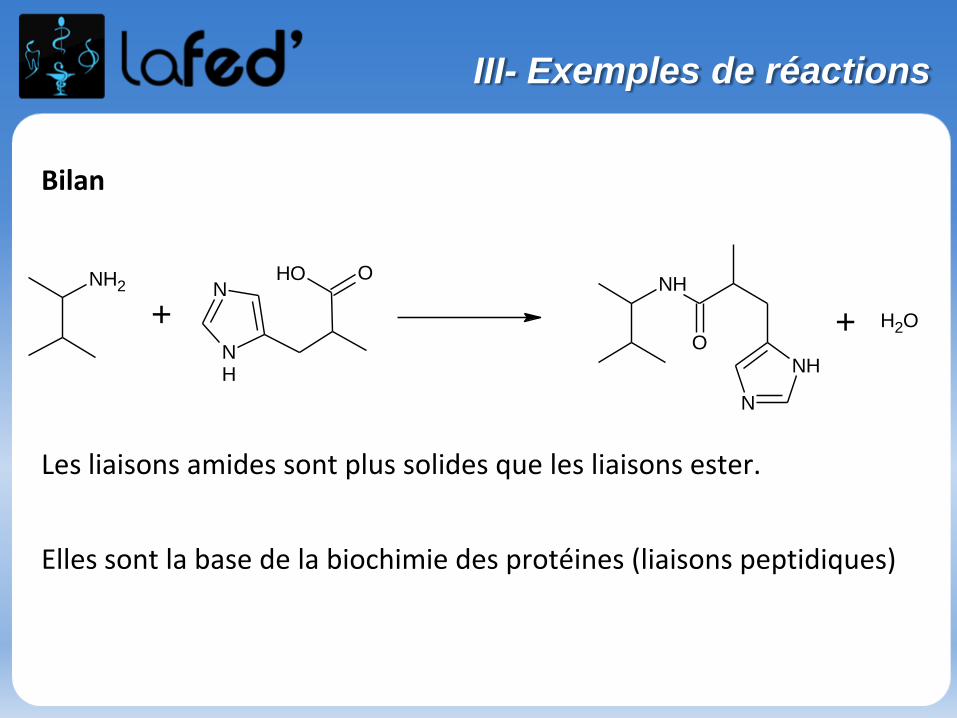
Bilan
Les liaisons amides sont plus solides que les liaisons ester.
Elles sont la base de la biochimie des protéines (liaisons peptidiques)
NH2
OH2+ +
OOH
NH
N NH
NH
N
O
III- Exemples de réactions

Réaction d’acétalisation/cétalisation :
Aldéhyde (ou cétone) + alcool → hémi-acétal (ou hémi-cétal)
Hémi-acétal (ou hémi-cétal) + alcool → acétal (ou cétal) + H2O
- Première étape = addition nucléophile
- Deuxième étape = substitution nucléophile
Etudions la réaction entre : Une cétone
Un alcool
O
OH
III- Exemples de réactions
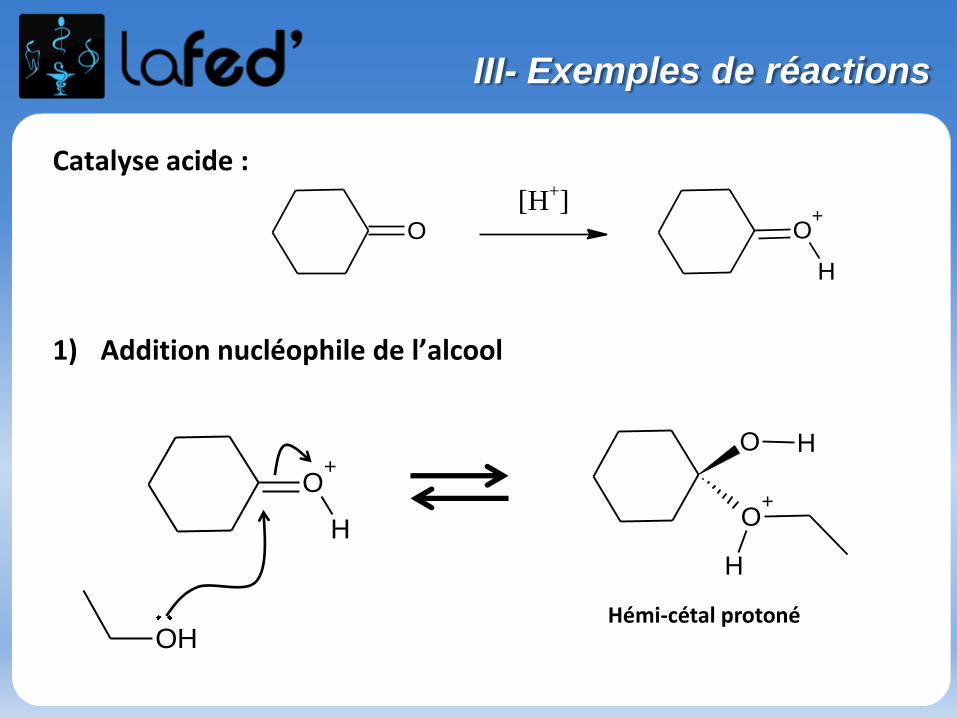
Catalyse acide :
1) Addition nucléophile de l’alcool
O
[H+]O
+
H
O+
H
OH
O H
O+
H
Hémi-cétal protoné
III- Exemples de réactions
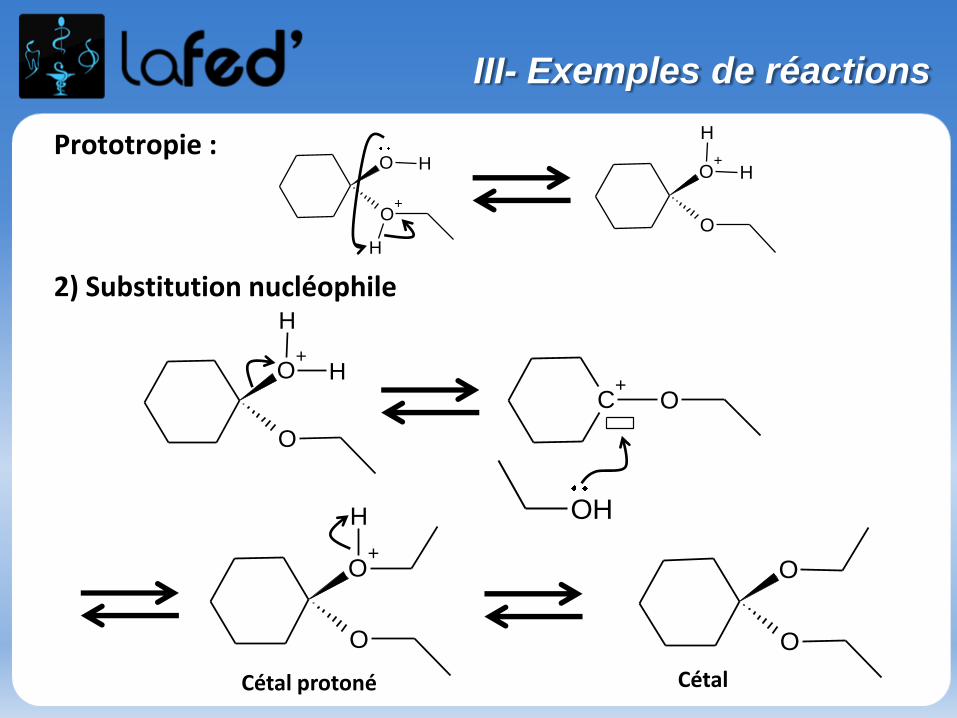
Prototropie :
2) Substitution nucléophile
O H
O+
H
O+
H
O
H
O+
H
O
H
C+
O
OH
O+
O
H
O
O
CétalCétal protoné
III- Exemples de réactions
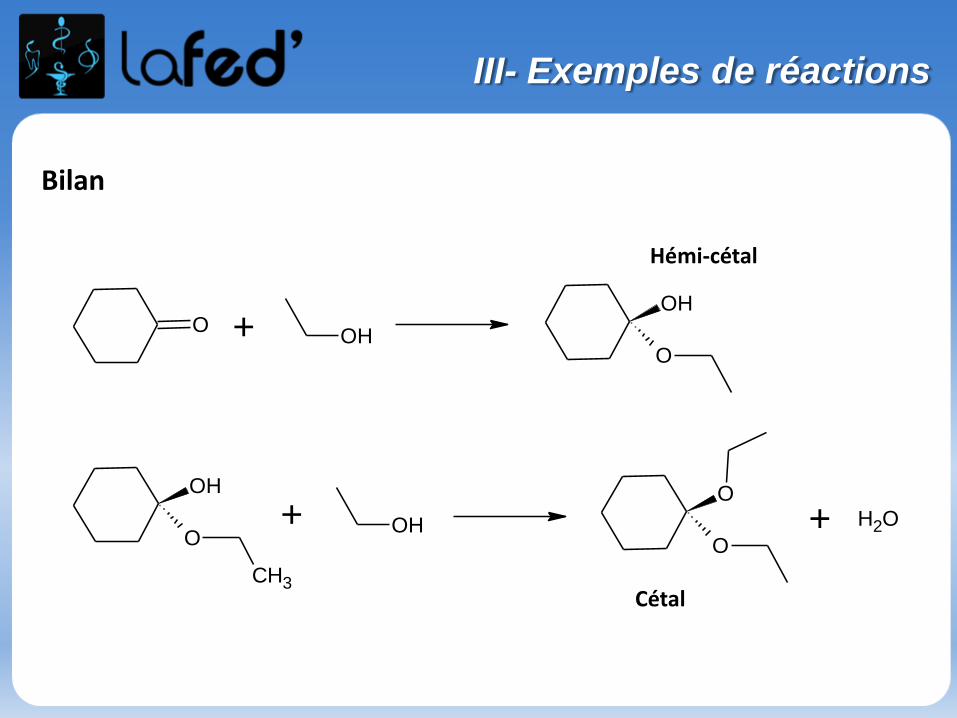
Bilan
O + OH
OH
O
OH
O
CH3
+ OH
O
O+ OH2
Cétal
Hémi-cétal
III- Exemples de réactions

Stage de Pré-Rentrée 2012UE 1 – Les protides
175
Diaporama réalisé par les tuteurs de La Fed’

176
• Sont appelés protides = acides aminés, peptides, protéines.
Les peptides et protéines sont des polymères d’acides aminés.
• Les protides sont des molécules du vivant azotées. On les retrouve dansl’alimentation. Leur dégradation produit de l’énergie.
- Protides (Viande rouge/Blancs d’œufs/Volaille)
- Glucides (sucres rapides/sucres lents)
- Lipides (huile, matières grasses, etc…)
• Les protides sont également des constituants structuraux essentiels ducorps humains (muscles +++) et fonctionnels (enzymes…).
10000 Daltons50 aaProtéines< 10000 Daltons< 50 aaPeptides
Poids moléculaire (PM)Nombre d’acides aminés
I- Généralités

Les protides peuvent être (non exhaustif !)
- des hormones ex: Insuline, FSH, Vasopressine…
- des enzymes ex: Glucokinase, ARN Polymérase…
- des unités structurales tissulaires ex: Collagène, Elastine…
- des transporteurs de molécules ex: Hémoglobine (transport O2)
- des molécules de reconnaissance ex: Immunoglobuline, Lectines…
- Des acteurs des mouvements mécaniques ex: Actine et Myosine,
Kinésine et Dinéine
Grande diversité structurale = Grande diversité fonctionnelle
I- Généralités

I- Généralités
Exercice Où sont donc cachés les protides ??

Les acides α-aminés sont des molécules chimiques constitués des atomes suivants:
C O H N S Se
Les AA possèdent tous, au moins :
1 Carbone central : Carbone Alpha
entouré de 4 substituants (tous différents sauf dans un cas GLYCINE)
→ 1 Fonction Amine (NH2)
→ 1 Fonction Acide carboxylique (COOH)
→ 1 Radical (variant selon l’acide aminé)
→ 1 hydrogène
1) Généralités sur les acides α-aminés
II- Les acides aminés
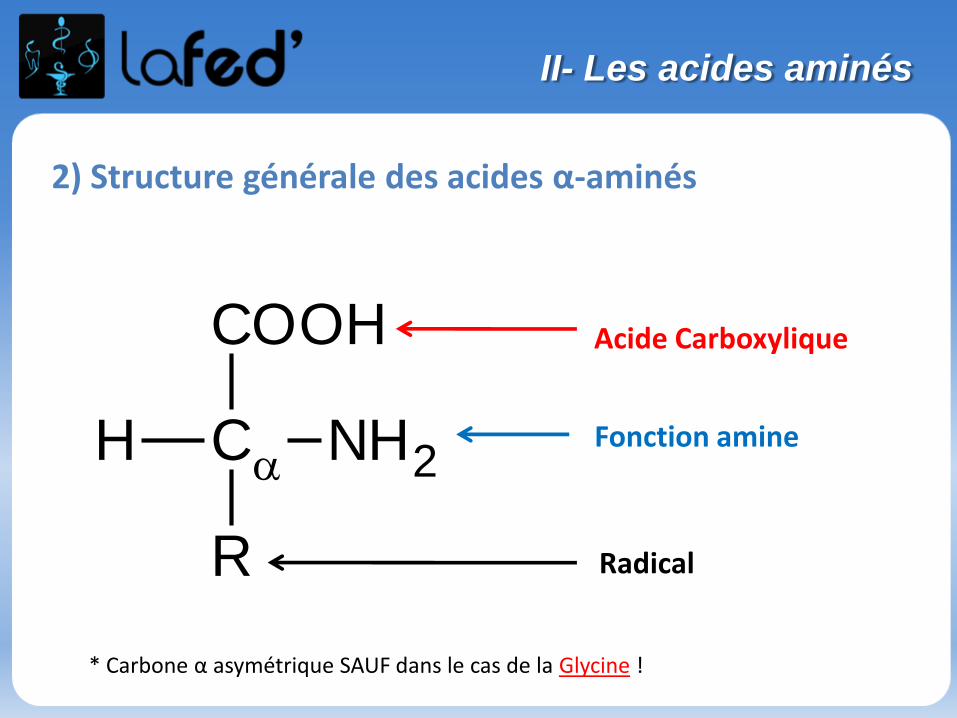
Acide Carboxylique
Fonction amine
Radical
COOH
C
R
H NH2
* Carbone α asymétrique SAUF dans le cas de la Glycine !
II- Les acides aminés
2) Structure générale des acides α-aminés
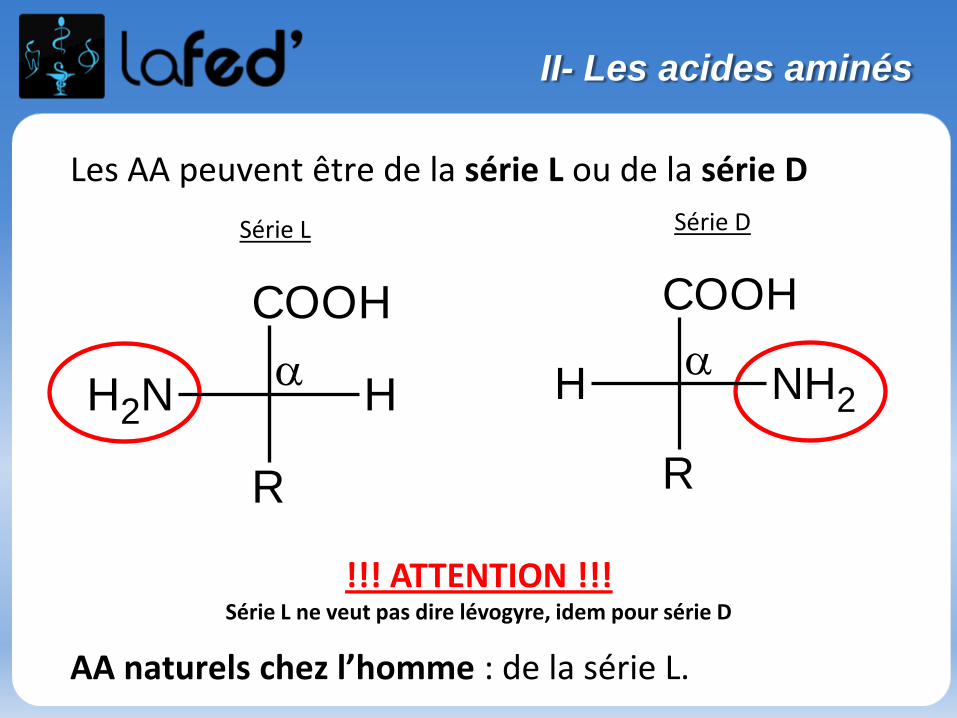
Les AA peuvent être de la série L ou de la série D
AA naturels chez l’homme : de la série L.
!!! ATTENTION !!! Série L ne veut pas dire lévogyre, idem pour série D
Série L Série D
II- Les acides aminés
COOH
R
NH2 H
COOH
R
H NH2

182
• Protéinogène : Acide aminé incorporé tel quel au cours du processus de traduction.
• 20 aa à retenir (+2 exotiques), classés dans différents groupes selon leur chaîne latérale (R)
A) Les aa à chaînes latérales non polaires, non chargées (9 aa)
Glycine, Gly, G Alanine, Ala, A
Valine, Val, V Leucine, Leu, L
Isoleucine, Ile, I Méthionine, Met, M
Proline, Pro, P Phénylalanine, Phe, F
Tryptophane, Trp, W
II- Les acides aminés
3) Acides aminés protéinogènes
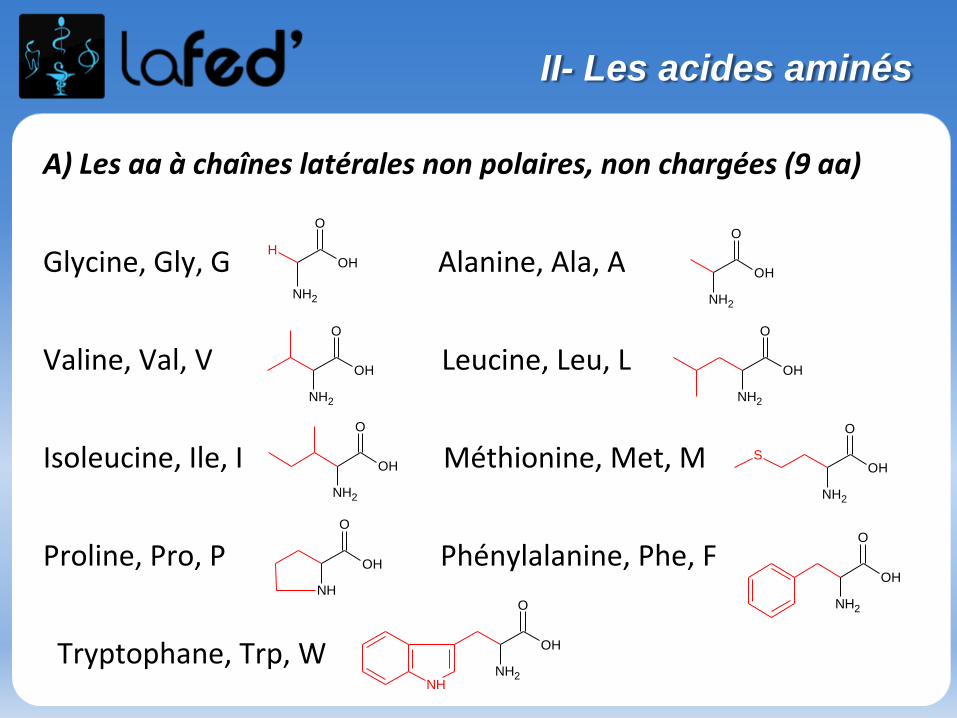
A) Les aa à chaînes latérales non polaires, non chargées (9 aa)
Glycine, Gly, G Alanine, Ala, A
Valine, Val, V Leucine, Leu, L
Isoleucine, Ile, I Méthionine, Met, M
Proline, Pro, P Phénylalanine, Phe, F
Tryptophane, Trp, W
O
NH2
OH
O
NH2
OH
O
NH2
OH
O
NH2
OH
O
NH2
SOH
O
NH2
OH
O
NH
OH
O
NH2NH
OH
II- Les acides aminés
O
NH2
OHH
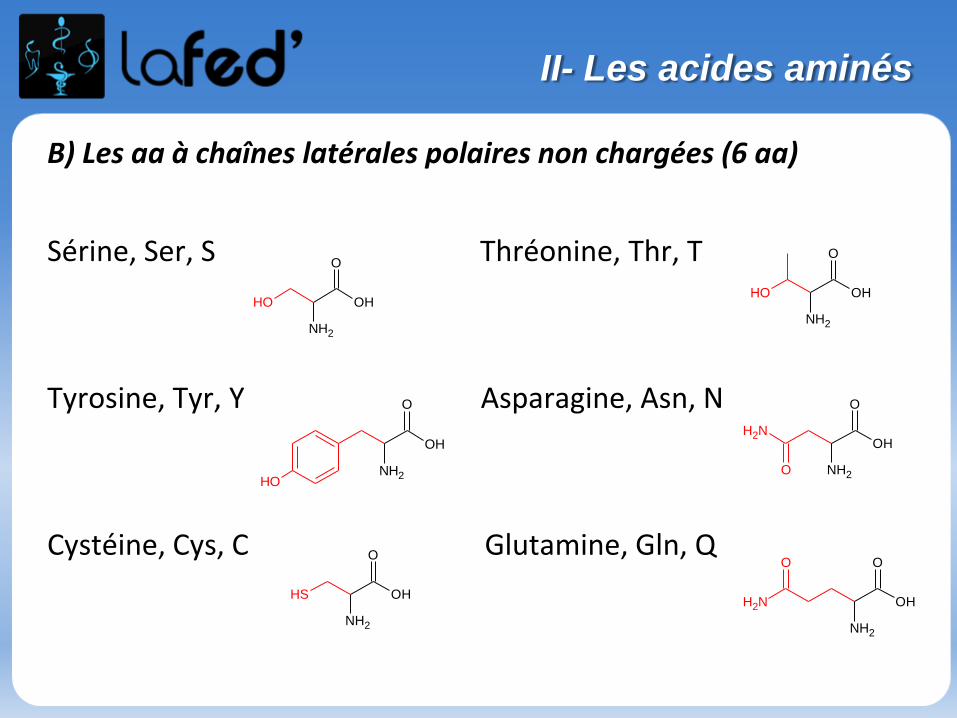
B) Les aa à chaînes latérales polaires non chargées (6 aa)
Sérine, Ser, S Thréonine, Thr, T
Tyrosine, Tyr, Y Asparagine, Asn, N
Cystéine, Cys, C Glutamine, Gln, Q
O
NH2
OH OH
O
NH2
OH OH
O
NH2
OH
OH
O
O
NH2
NH2
OH
O
NH2
O
NH2
OH
O
NH2
SH OH
II- Les acides aminés
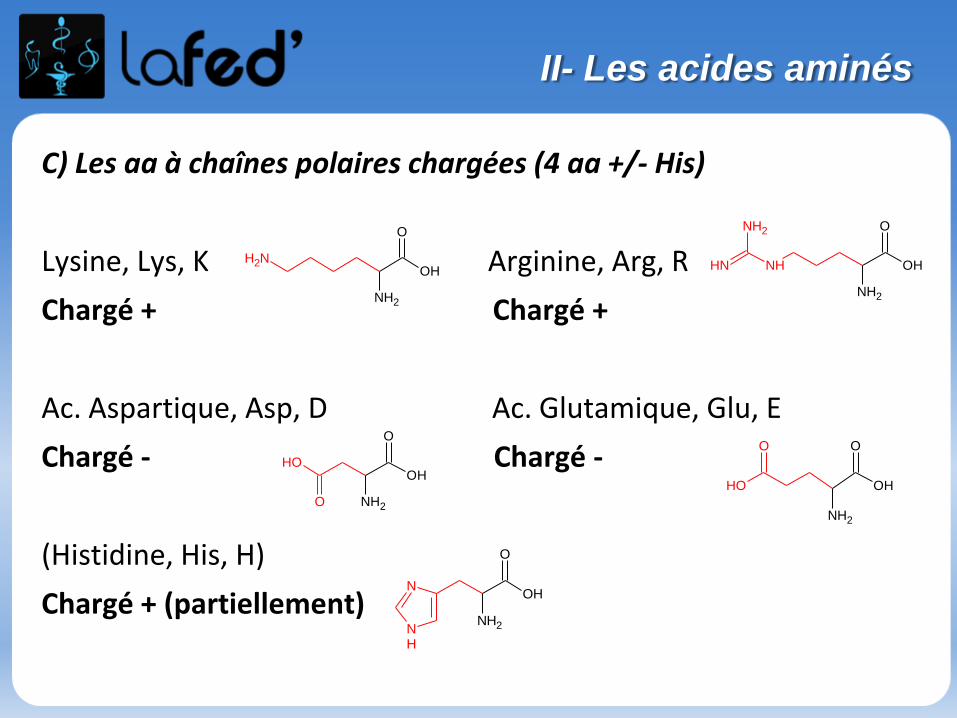
C) Les aa à chaînes polaires chargées (4 aa +/- His)
Lysine, Lys, K Arginine, Arg, R
Chargé + Chargé +
Ac. Aspartique, Asp, D Ac. Glutamique, Glu, E
Chargé - Chargé -
(Histidine, His, H)
Chargé + (partiellement)
O
NH2
NH2OH NH
O
NH
NH2
NH2
OH
O O
OHOH
NH2
O
O
OH
NH2
OH
O
NH2
N
NH
OH
II- Les acides aminés
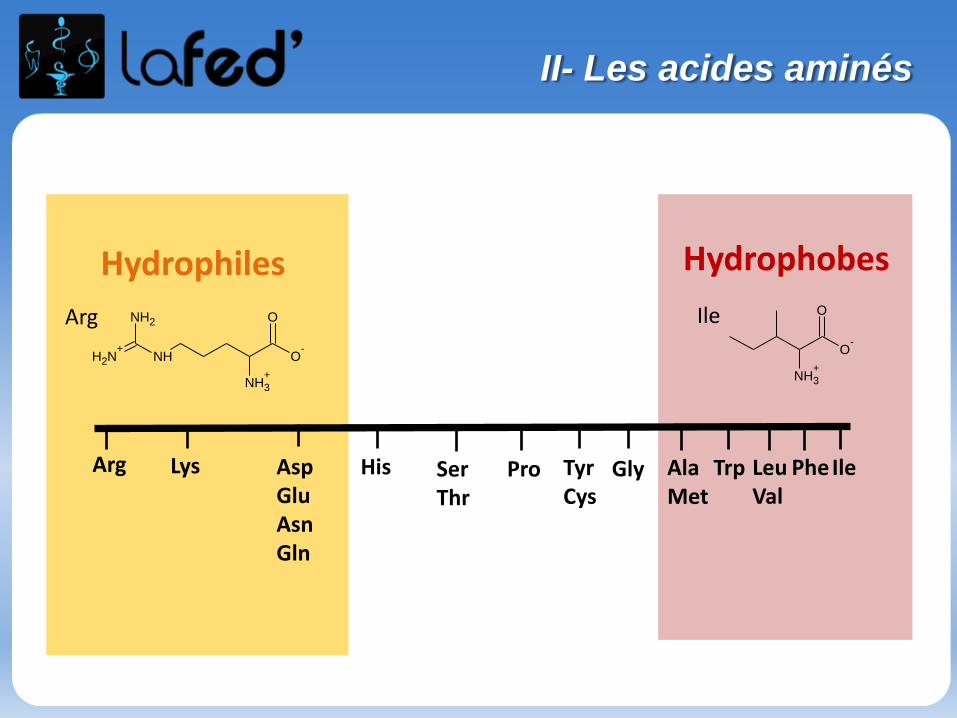
Arg Lys AspGluAsnGln
His SerThr
Pro TyrCys
Gly AlaMet
Trp PheIleLeuVal
Hydrophiles Hydrophobes
NH2
+
O
NH
NH2
NH3
+
O-
O
NH3
+
O-
Arg Ile
II- Les acides aminés
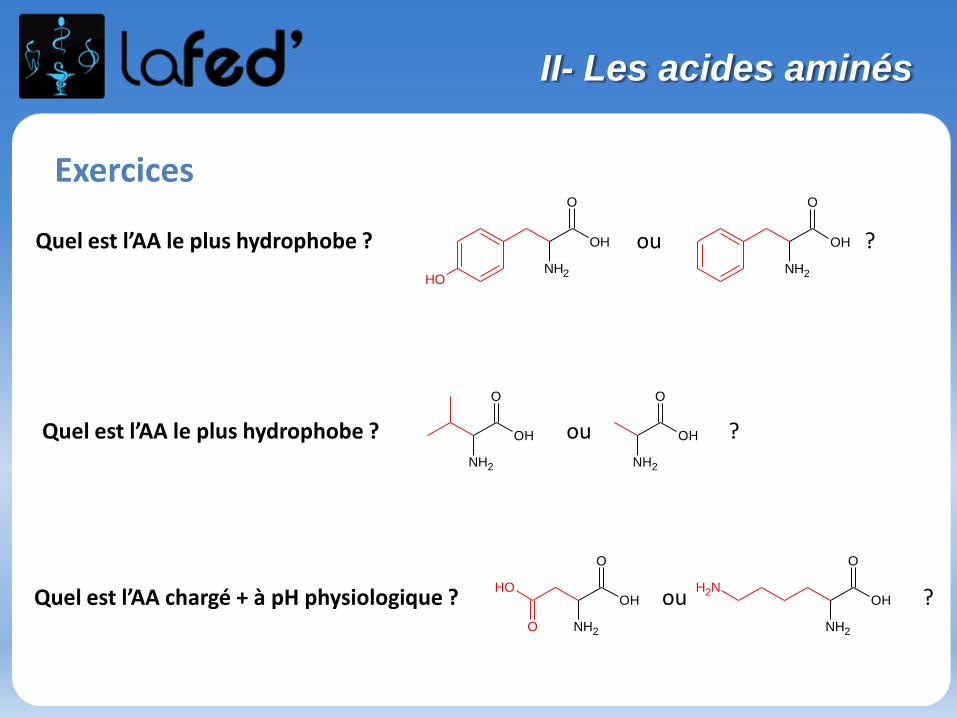
Exercices
Quel est l’AA le plus hydrophobe ? ou ?
O
NH2
OH
OH
O
NH2
OH
Quel est l’AA chargé + à pH physiologique ? ou ?
O
O
OH
NH2
OH
Quel est l’AA le plus hydrophobe ? ou ?
O
NH2
OH
O
NH2
OH
O
NH2
NH2OH
II- Les acides aminés

Exercices
Parmi ces acides aminés, lequel peut former des ponts dissulfures S-S lorsqu’il est oxydé ?
Sérine
Cystéine
Methionine
Phénylalanine
Tout le monde veut prendre sa place (En P2 !)
II- Les acides aminés
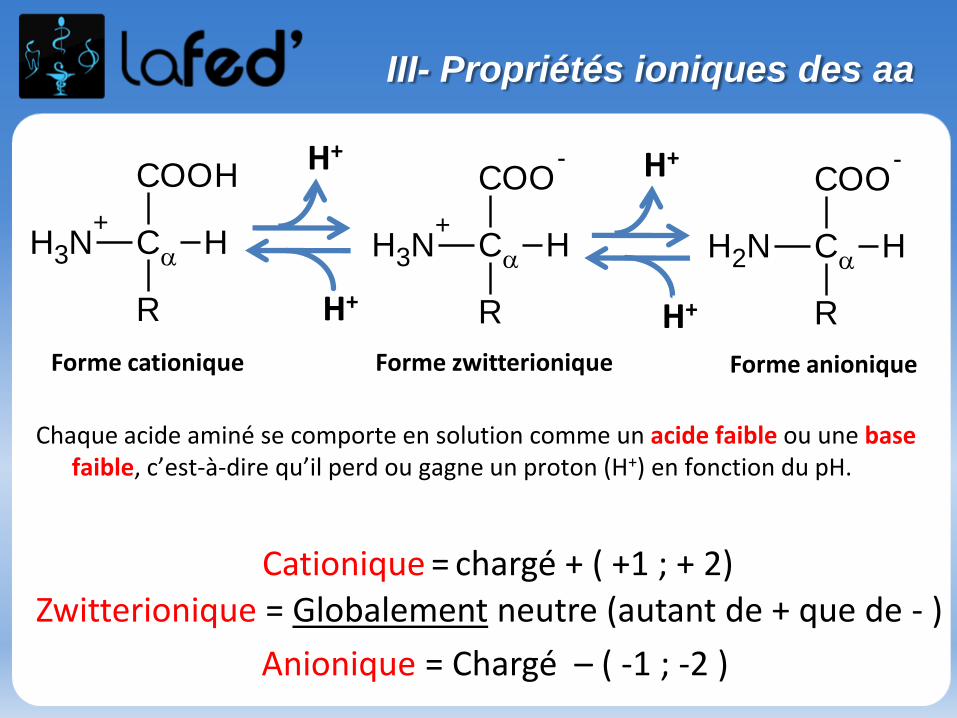
Cationique = chargé + ( +1 ; + 2)Zwitterionique = Globalement neutre (autant de + que de - )
Anionique = Chargé – ( -1 ; -2 )
COOH
C
R
NH3
+H
COO-
C
R
NH3
+H
COO-
C
R
NH2 H
H+H+
H+ H+
Forme cationique Forme zwitterionique Forme anionique
Chaque acide aminé se comporte en solution comme un acide faible ou une base faible, c’est-à-dire qu’il perd ou gagne un proton (H+) en fonction du pH.
III- Propriétés ioniques des aa

Pour modéliser correctement ces phénomènes, nous avons besoin de notions de chimie physique.
Nous allons donc faire un saut dans le passé, en Terminale S…
III- Propriétés ioniques des aa

• D’après Brønsted (1923) :
Un acide est une entité capable de libérer un ion H+.
On écrit : AH+ → A + H+ ou bien AH → A- + H+
Une base est une entité capable de capturer un ion H+.
On écrit : B + H+ → BH+ ou bien B- + H+ → BH
III- Propriétés ioniques des aa

• Les réactions peuvent se dérouler dans les deuxsens, en fonction des conditions du milieu.
• Conséquence : A chaque acide est associé unebase conjuguée, et vice-versa.
AH+ ↔ A + H+
acide base conjuguée
Couple acido basique écrit par convention : (AH+/A)(Acide/Base)
III- Propriétés ioniques des aa
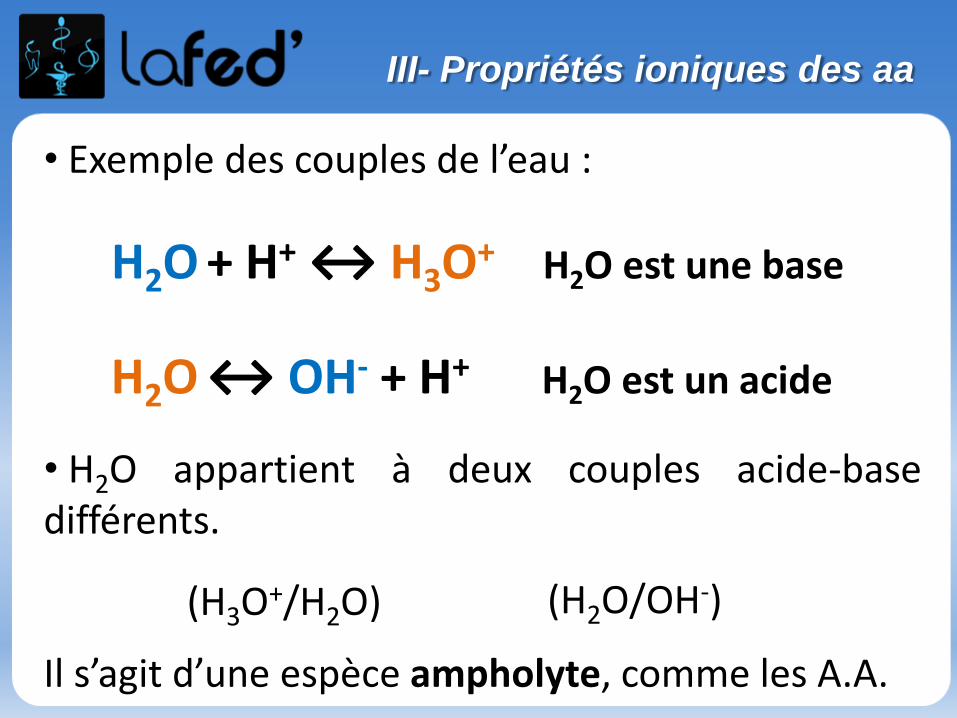
• Exemple des couples de l’eau :
H2O + H+ ↔ H3O+ H2O est une base
H2O ↔ OH- + H+ H2O est un acide
• H2O appartient à deux couples acide-basedifférents.
Il s’agit d’une espèce ampholyte, comme les A.A.
(H3O+/H2O) (H2O/OH-)
III- Propriétés ioniques des aa

• Soit la réaction acido-basique :
AH+ + B- ↔ A + BH
• La thermodynamique apporte des réponses auxquestions suivantes :
- Dans quel sens la réaction se déroule-t-elle ?- La réaction est-elle équilibrée ou totale ?
• Cela est fonction de la « force » relative des couplesacide-base mis en jeu. Il nous faut un critèrethermodynamique pour classer ces différents couples.
III- Propriétés ioniques des aa

• Recherchons la constante d’équilibre K de laréaction AH+ + B- ↔ A + BH
Si à t0 tous les réactants sont à concentration équimolaire :
K > 1, la réaction se déroule de gauche à droite (noté sens 1).K < 1, la réaction se déroule de droite à gauche (noté sens 2 ou -1).
K très grand ou très faible : réaction totale.
(A)eq.(BH)eq
(AH+)eq.(B-)eq
K =
III- Propriétés ioniques des aa

• La détermination expérimentale de K pourchaque réaction acido-basique est fastidieuse…
• On préfère comparer chaque couple acide-base àun couple de référence.
Couple de référence choisi : H3O+/H2O
• En vue d’établir un classement des acides selonleur force.
III- Propriétés ioniques des aa

• On détermine, pour un couple donné, la constanted’acidité KA.
• Il s’agit de la constante d’équilibre de la réaction de laforme acide avec l’eau. (facilité de la protonation de H2O)
• Réaction : AH+ + H2O ↔ A + H3O+
• Expression de KA Solution aqueuse diluée
(A)eq.(H3O+)eq
(AH+)eq.(H2O)eq
KA =[A]eq.[H3O
+]eq
[AH+]eq
KA =
III- Propriétés ioniques des aa

• KA, comme toute constante d’équilibre K, ne dépend quede la température.
• Les valeurs de Ka peuvent être très grandes ou trèsfaibles. Pour comparer l’acidité, on définit le pKA.
pKA = - log (KA)
• Et bien évidemment :
KA = 10 -pKa
III- Propriétés ioniques des aa

• En 1909, Sørensen définit le potentiel hydrogène (ou pH)
pH = - log (H3O+)
NB: En solution aqueuse diluée, (H3O+) ≈ [H3O+]
• Le pH d’une solution s’étend de 0 à 14.
pH = 7,0 solution neutre [H3O+] = [OH-]pH < 7,0 solution acide [H3O+] > [OH-]pH > 7,0 solution basique [H3O+] < [OH-]
III- Propriétés ioniques des aa
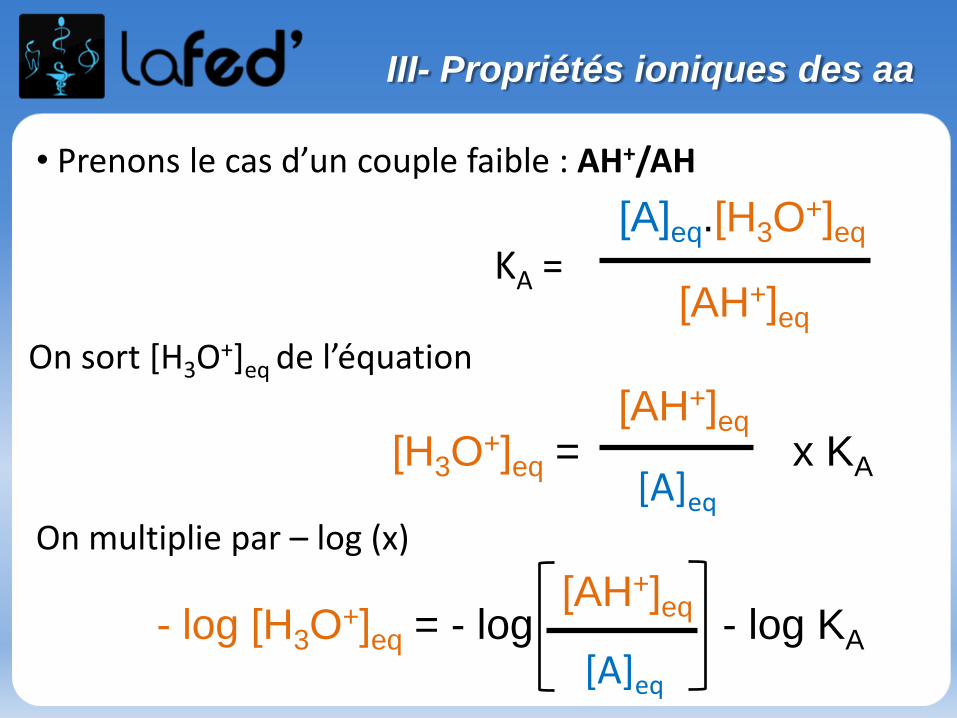
- log [H3O+]eq = - log - log KA
• Prenons le cas d’un couple faible : AH+/AH
[A]eq.[H3O+]eq
[AH+]eq
KA =
[A]eq
[AH+]eq
[H3O+]eq = x KA
On sort [H3O+]eq de l’équation
[A]eq
[AH+]eq
On multiplie par – log (x)
III- Propriétés ioniques des aa
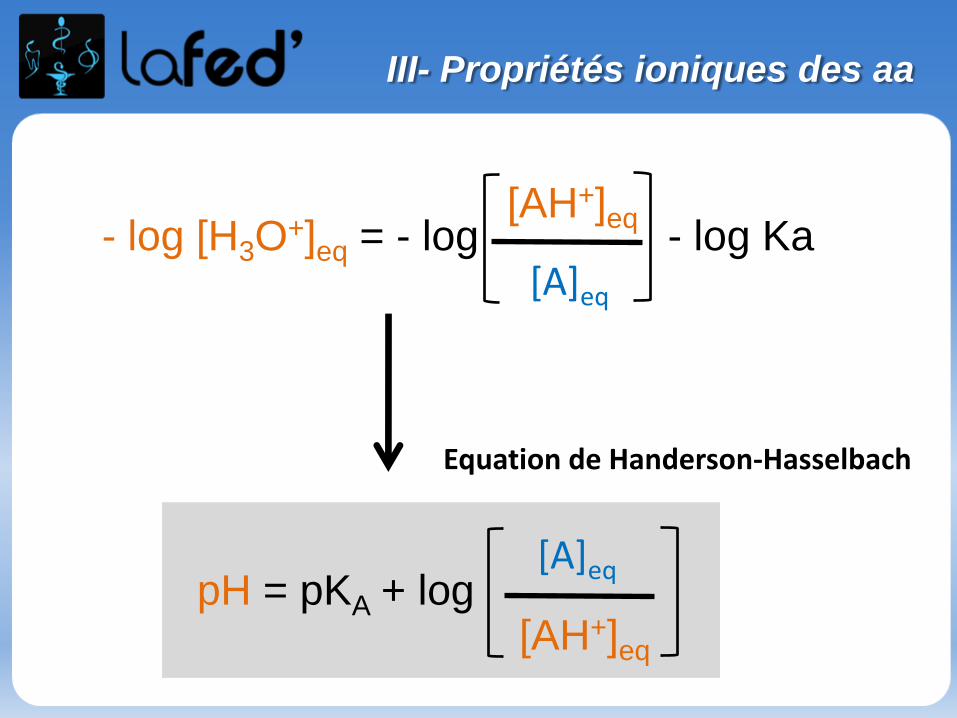
pH = pKA + log[A]eq
[AH+]eq
- log [H3O+]eq = - log - log Ka
[A]eq
[AH+]eq
Equation de Handerson-Hasselbach
III- Propriétés ioniques des aa

A pH = pKA , on a log (A/AH+) = 0et donc A/AH+ = 1 : demi-dissociation
pH = pKA + log[A]eq
[AH+]eq
III- Propriétés ioniques des aa
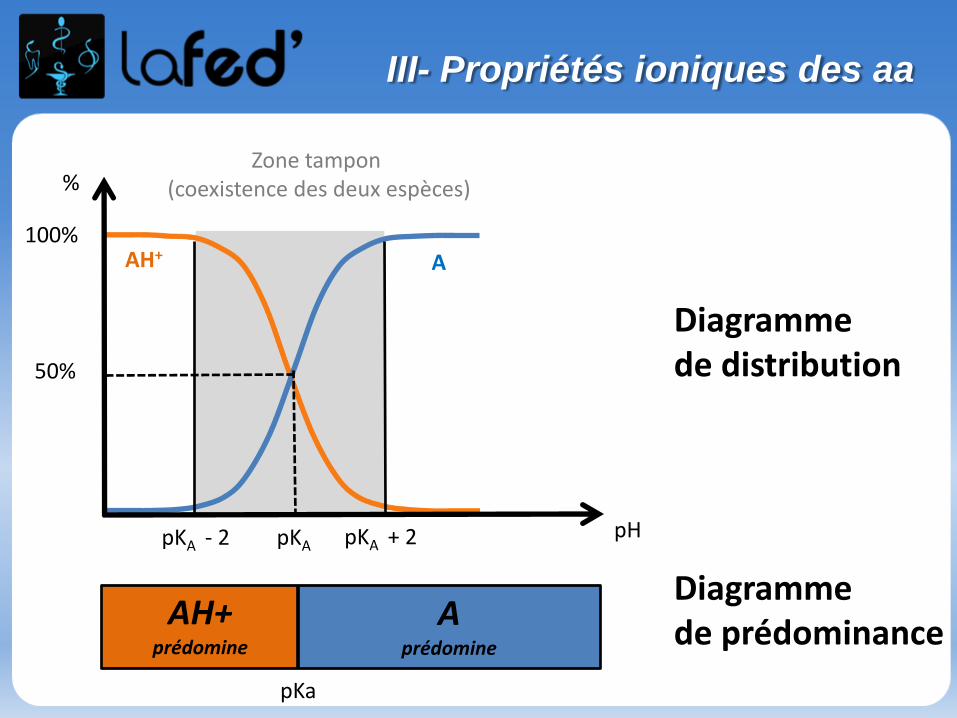
Diagrammede distribution
Diagrammede prédominance
%
100%
50%
pHpKA
Zone tampon (coexistence des deux espèces)
pKA - 2 pKA + 2
AH+ A
AH+prédomine
Aprédomine
pKa
III- Propriétés ioniques des aa
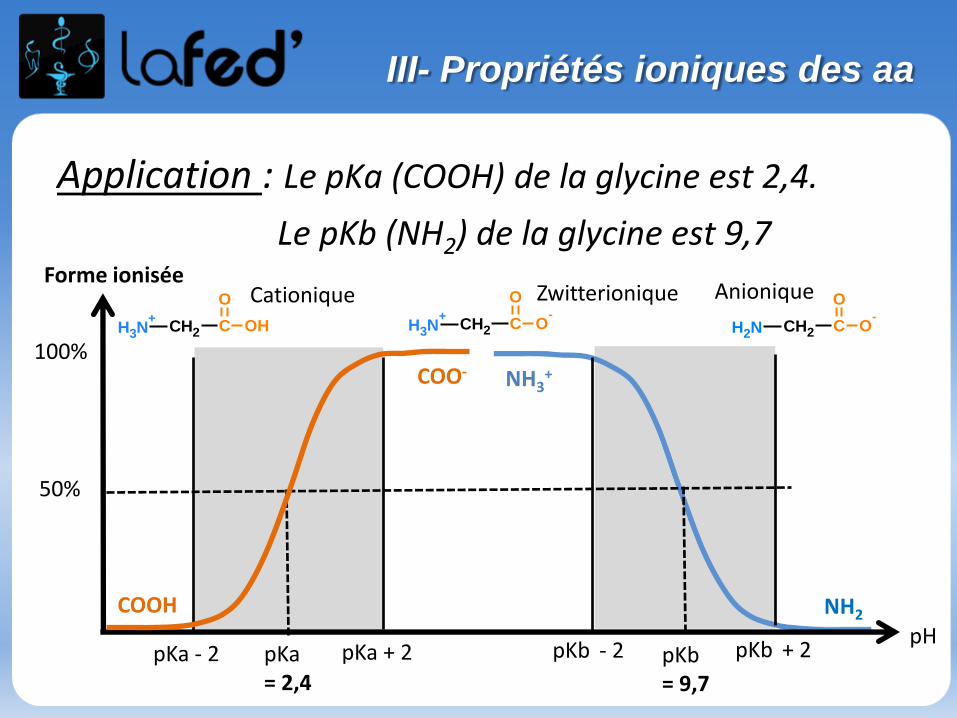
Application : Le pKa (COOH) de la glycine est 2,4.
Le pKb (NH2) de la glycine est 9,7 Forme ionisée
100%
50%
pHpKa= 2,4
pKa - 2 pKa + 2
COO-
NH2
NH3+
pKb - 2 pKb + 2
COOH
C
O
NH3
+CH2 O
-
pKb= 9,7
C
O
NH2CH2 O
-C
O
NH3
+CH2 OH
Cationique Zwitterionique Anionique
III- Propriétés ioniques des aa
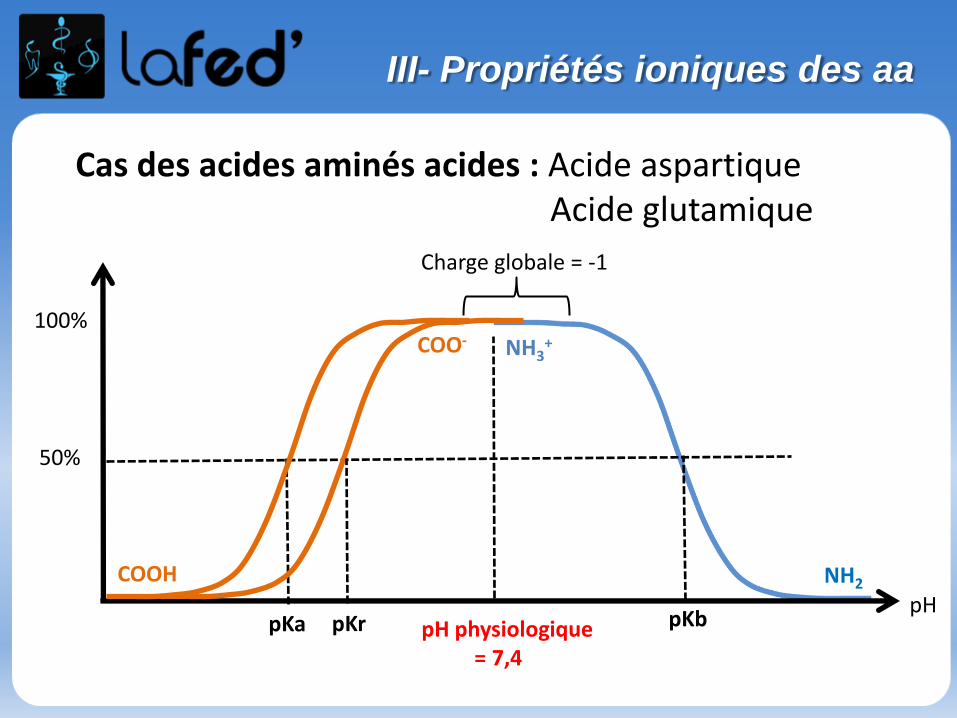
100%
50%
pH
COO-
NH2
NH3+
COOH
Cas des acides aminés acides : Acide aspartiqueAcide glutamique
Charge globale = -1
pKa pKr pKbpH physiologique= 7,4
III- Propriétés ioniques des aa

100%
50%
pH
COO-
NH2
NH3+
COOH
Cas des acides aminés basiques : LysineArginine (+ Histidine)
Charge globale = + 1
pKa pKbpH physiologique pKr
III- Propriétés ioniques des aa
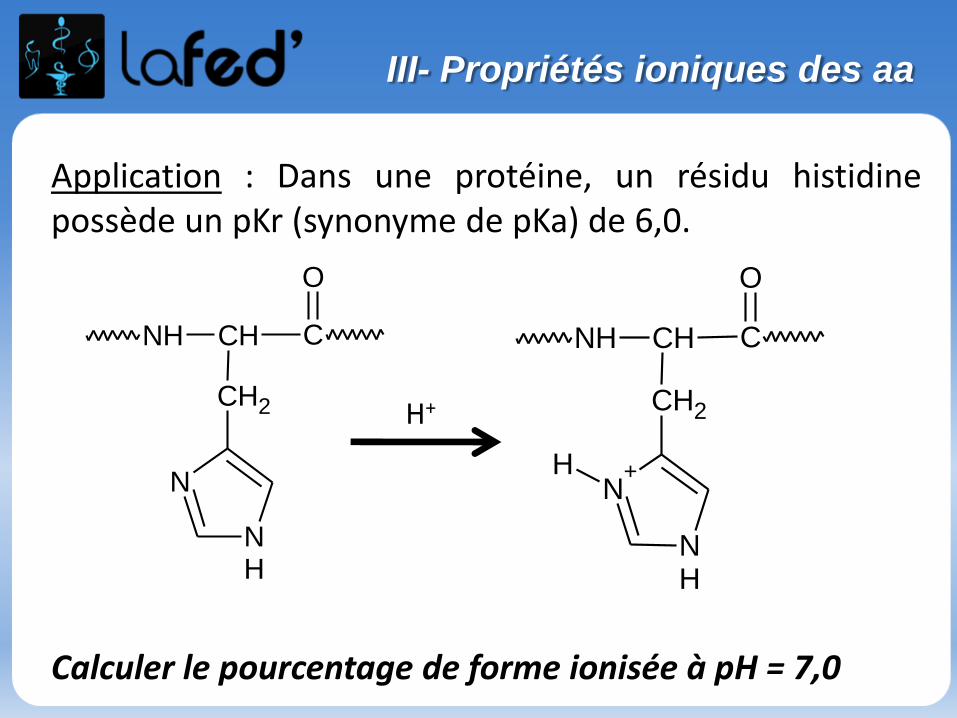
Application : Dans une protéine, un résidu histidinepossède un pKr (synonyme de pKa) de 6,0.
Calculer le pourcentage de forme ionisée à pH = 7,0
NH CH C
O
CH2
NH
NH
NH CH C
O
CH2
NH
N+
H+
III- Propriétés ioniques des aa
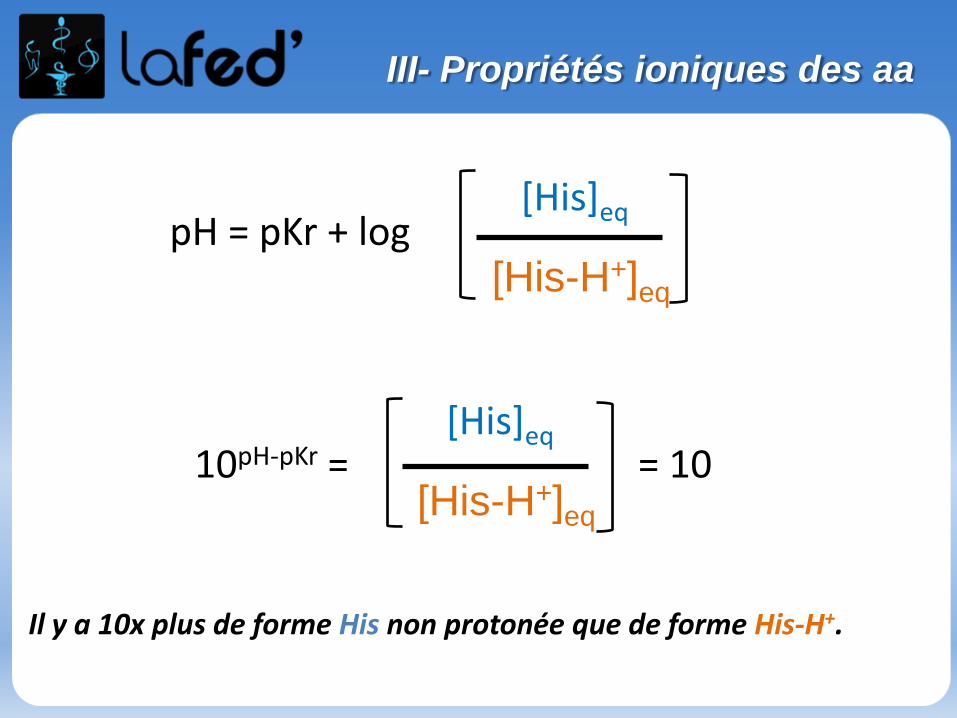
Il y a 10x plus de forme His non protonée que de forme His-H+.
pH = pKr + log[His]eq
[His-H+]eq
10pH-pKr = = 10[His]eq
[His-H+]eq
III- Propriétés ioniques des aa
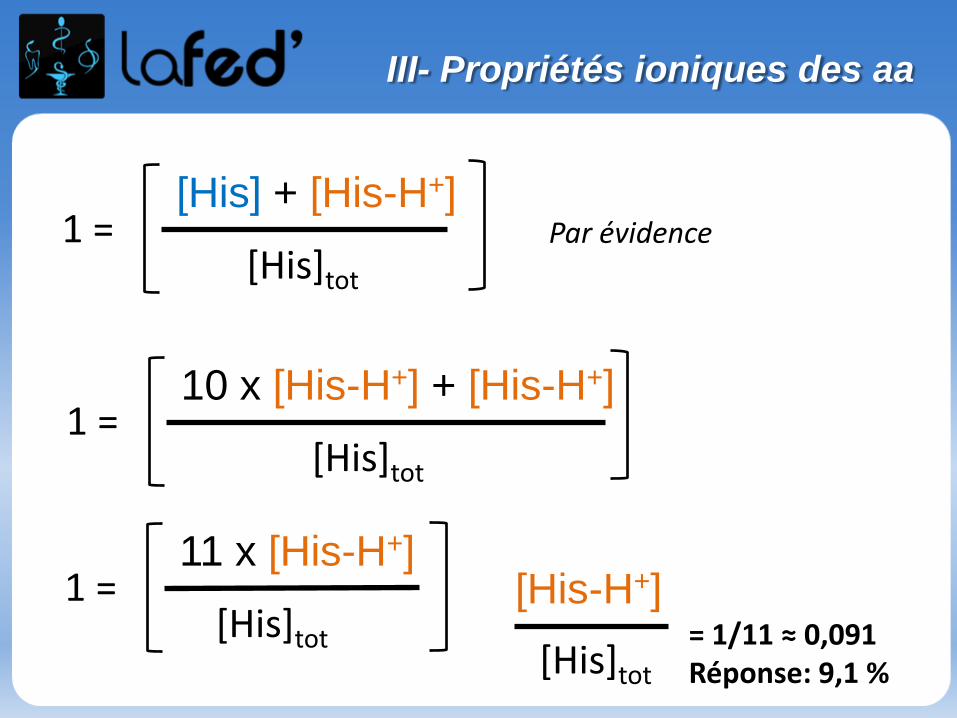
1 = [His] + [His-H+]
[His]tot
1 = 10 x [His-H+] + [His-H+]
[His]tot
1 = 11 x [His-H+]
[His]tot
[His-H+]
[His]tot
= 1/11 ≈ 0,091Réponse: 9,1 %
Par évidence
III- Propriétés ioniques des aa
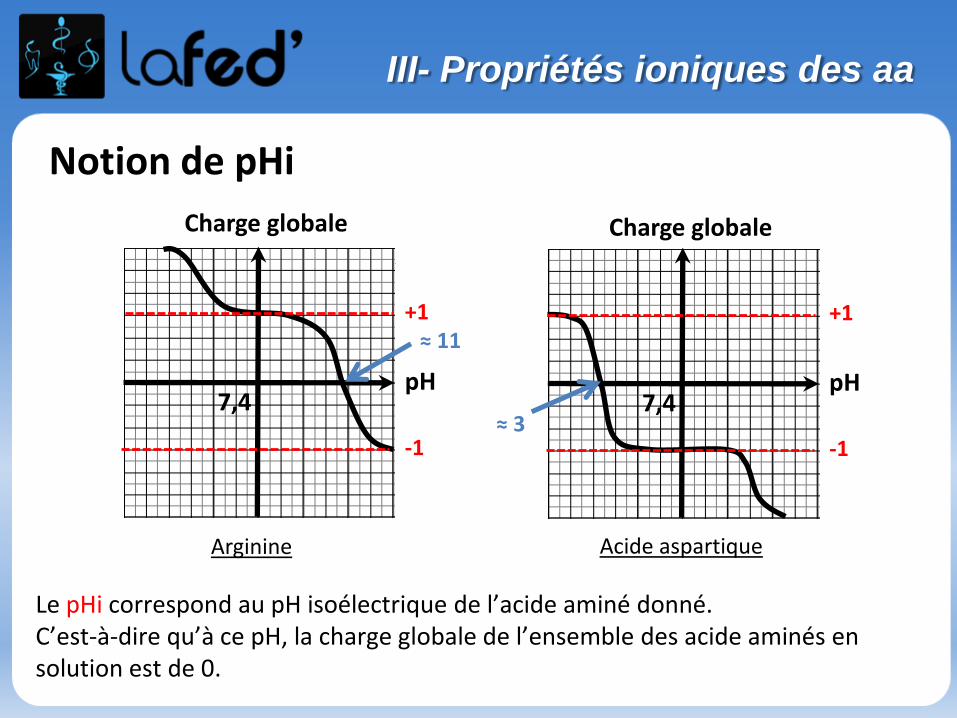
pH
Charge globale
+1
-1
7,4pH
Charge globale
+1
-1
7,4
Notion de pHi
Le pHi correspond au pH isoélectrique de l’acide aminé donné. C’est-à-dire qu’à ce pH, la charge globale de l’ensemble des acide aminés en solution est de 0.
Arginine Acide aspartique
≈ 3
≈ 11
III- Propriétés ioniques des aa
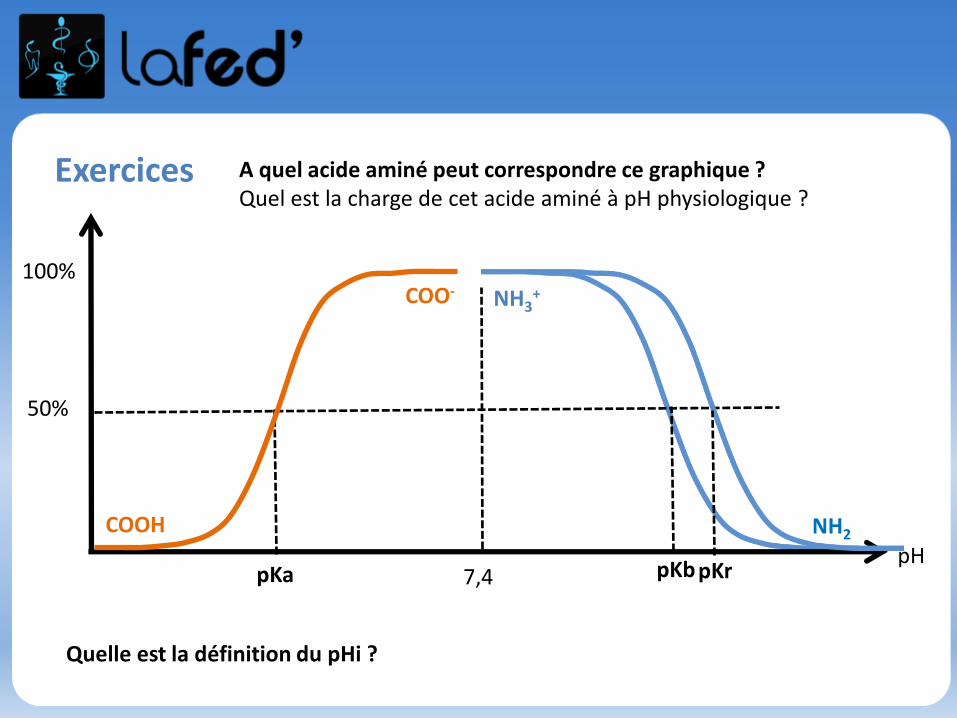
100%
50%
pH
COO-
NH2
NH3+
COOH
pKa pKbpKr
Exercices A quel acide aminé peut correspondre ce graphique ?Quel est la charge de cet acide aminé à pH physiologique ?
7,4
Quelle est la définition du pHi ?

Un peptide ou une protéine est un enchaînement linéaire d’une séquence d’acide aminés.
Tous les acides aminés peuvent s’enchaîner dans n’importe quel ordre donc beaucoup de possibilités d’enchaînements polypeptidiques.
IV- Peptides et protéines
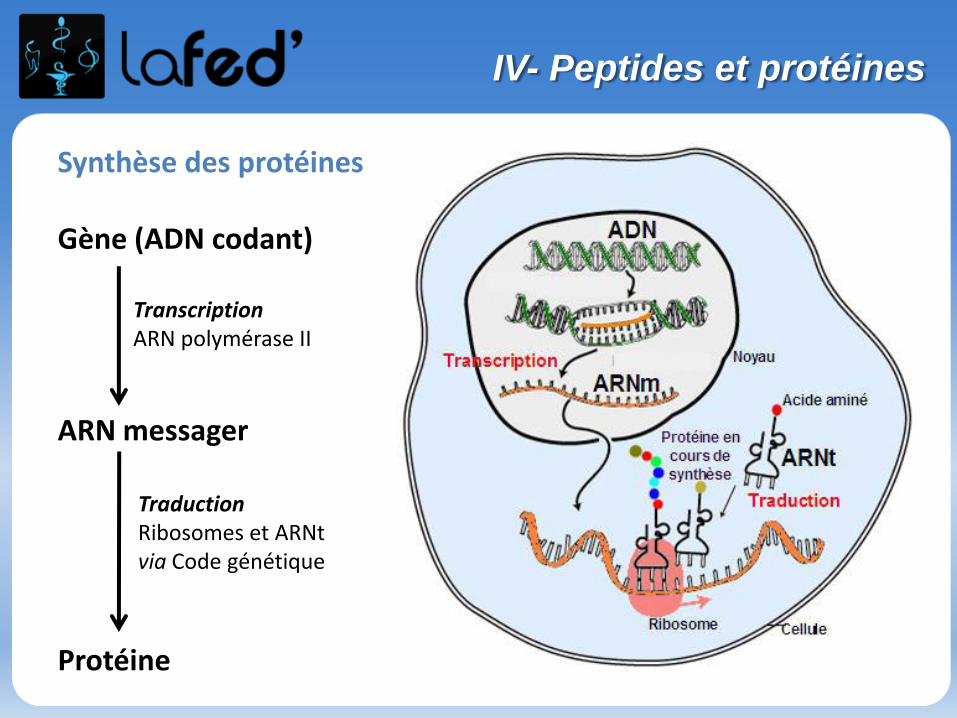
Synthèse des protéines
Gène (ADN codant)
ARN messager
Protéine
TraductionRibosomes et ARNtvia Code génétique
TranscriptionARN polymérase II
IV- Peptides et protéines
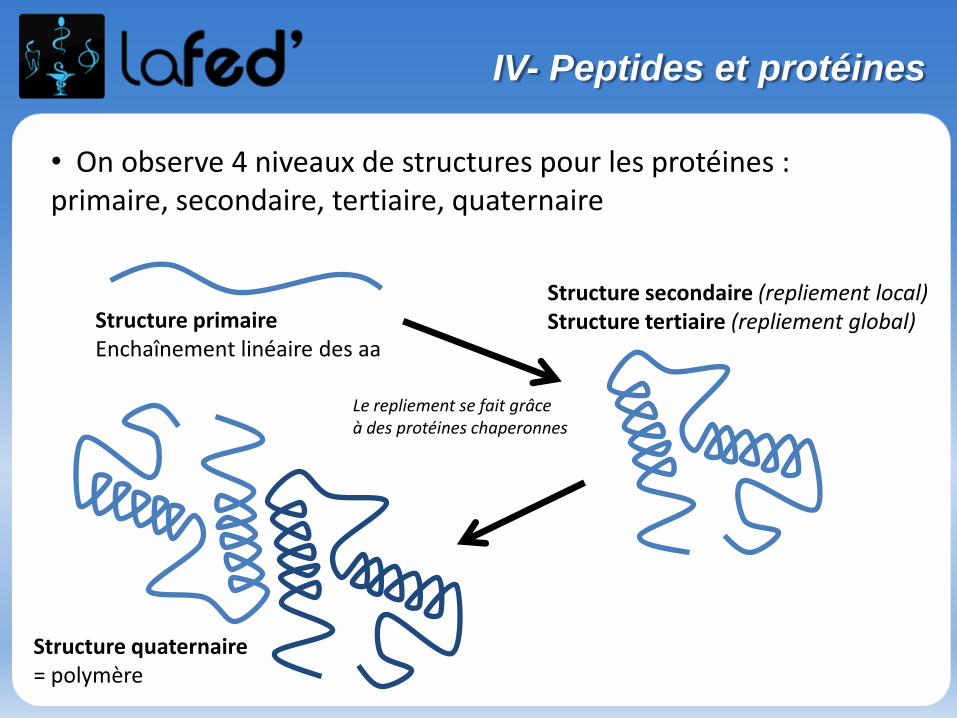
• On observe 4 niveaux de structures pour les protéines : primaire, secondaire, tertiaire, quaternaire
Structure primaireEnchaînement linéaire des aa
Structure secondaire (repliement local)Structure tertiaire (repliement global)
Structure quaternaire= polymère
Le repliement se fait grâce à des protéines chaperonnes
IV- Peptides et protéines
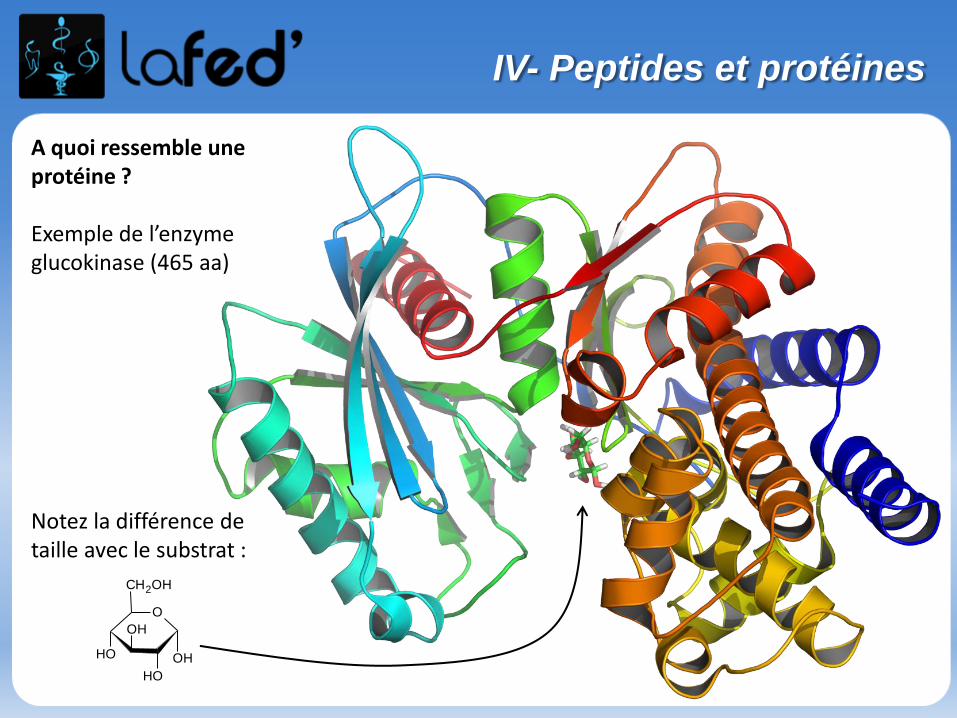
A quoi ressemble une protéine ?
Exemple de l’enzyme glucokinase (465 aa)
Notez la différence de taille avec le substrat :
O
OH
OH
OH OH
CH2OH
IV- Peptides et protéines

216
Enchaînement linéaire d’acides aminés, sans conformation particulière dans l’espace.
L’enchaînement est possible grâce a des liaisons peptidiques entre chaque acide aminé.
IV- Peptides et protéines
1) Structure primaire
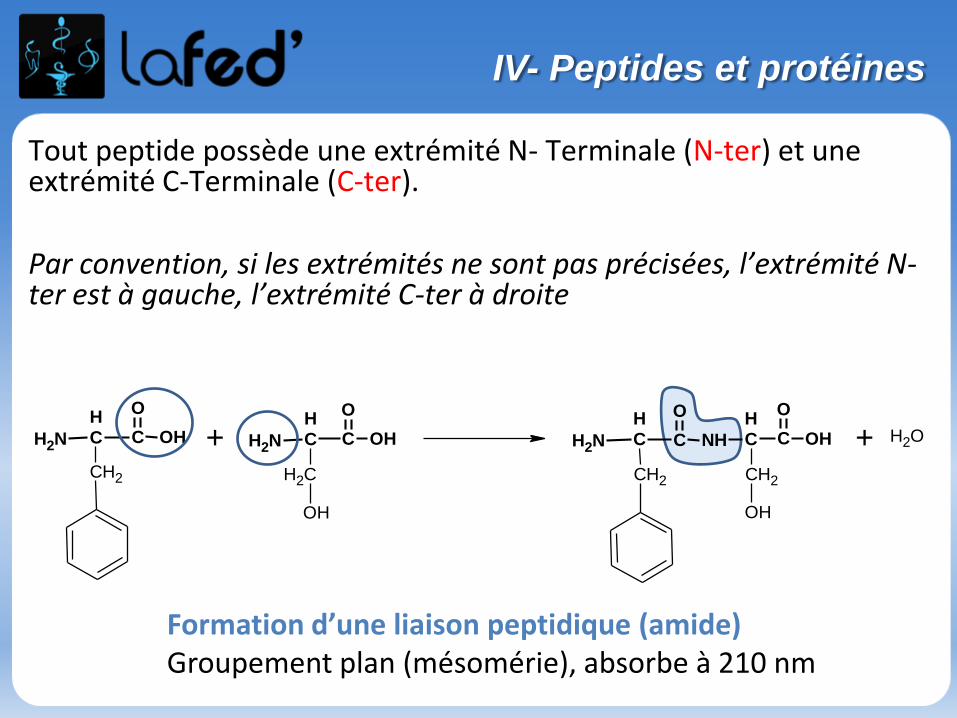
Tout peptide possède une extrémité N- Terminale (N-ter) et une extrémité C-Terminale (C-ter).
Par convention, si les extrémités ne sont pas précisées, l’extrémité N-ter est à gauche, l’extrémité C-ter à droite
C
O
NH2CH
OH
CH2
+ C
O
NH2CH
OH
CH2
OH
C
O
NH2CH
NH C
O
CH
OH
CH2 CH2
OH
+ OH2
Formation d’une liaison peptidique (amide)Groupement plan (mésomérie), absorbe à 210 nm
IV- Peptides et protéines

Modifications post-traductionnelles
Elles sont le fait d’enzymes qui modifient chimiquement la structure primaire. Il en existerait plus de 200 :
- Phosphorylation/déphosphorylation par des kinases et phosphatases. Concerne les résidus Tyr, Thr, Ser.
- Acétylation sur Lys (acétylation des protéines histones)
- Hydroxylation sur Pro (collagène)
- O-Glycosylation sur Thr/Ser
- N-Glycosylation sur Asn (séquence consensus N-X-S ou N-X-T)
- Excision du Met en N-ter à l’issue de la traduction
…
IV- Peptides et protéines

Structure permise grâce aux contraintes physiques et électroniques qu’exercent les résidus entre eux.
Donne des propriétés géométriques à l’enchaînement d’acides aminés
La structure secondaire est un premier « repli » de l’enchaînement d’acides aminés
IV- Peptides et protéines
2) Structure secondaire
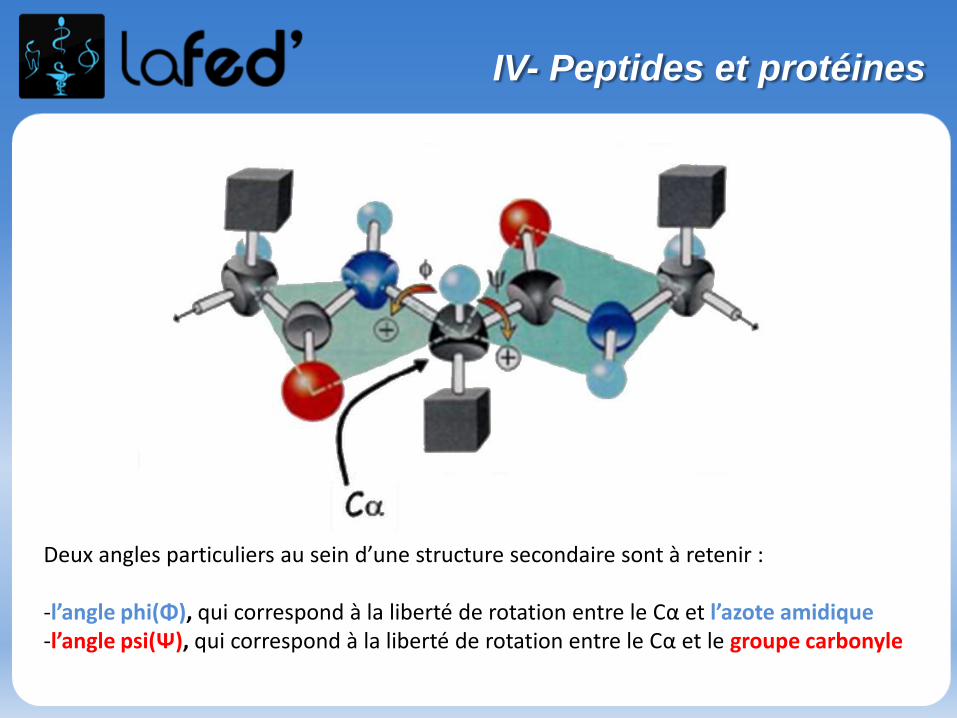
Deux angles particuliers au sein d’une structure secondaire sont à retenir :
-l’angle phi(Φ), qui correspond à la liberté de rotation entre le Cα et l’azote amidique-l’angle psi(Ψ), qui correspond à la liberté de rotation entre le Cα et le groupe carbonyle
IV- Peptides et protéines
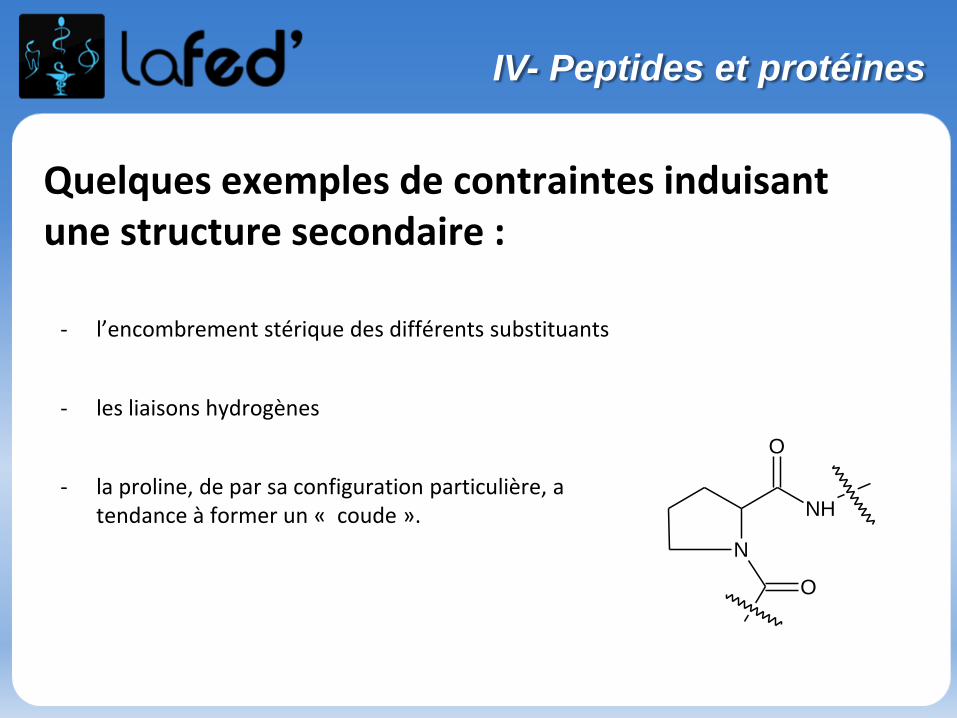
Quelques exemples de contraintes induisant une structure secondaire :
O
N
NH
O
- l’encombrement stérique des différents substituants
- les liaisons hydrogènes
- la proline, de par sa configuration particulière, a tendance à former un « coude ».
IV- Peptides et protéines
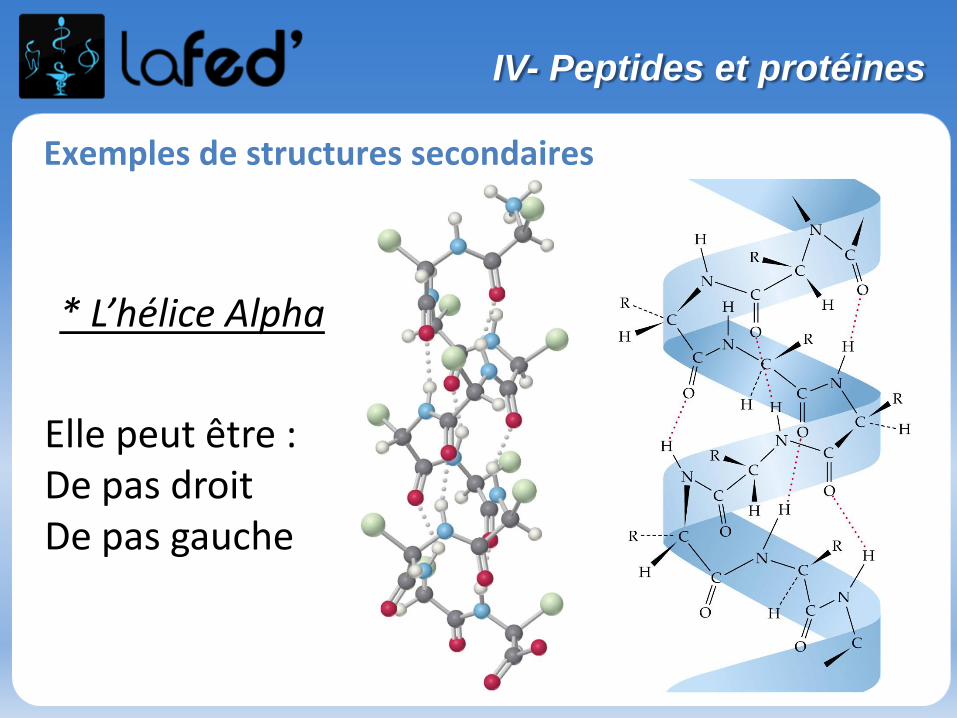
Exemples de structures secondaires
* L’hélice Alpha
Elle peut être :De pas droitDe pas gauche
IV- Peptides et protéines
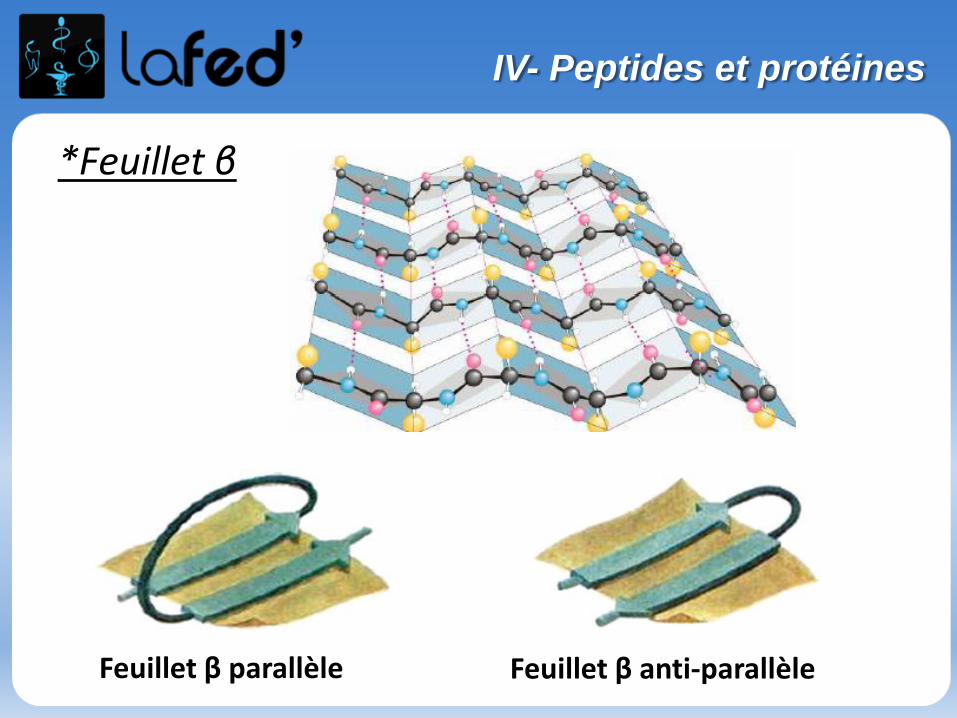
*Feuillet β
Feuillet β parallèle Feuillet β anti-parallèle
IV- Peptides et protéines
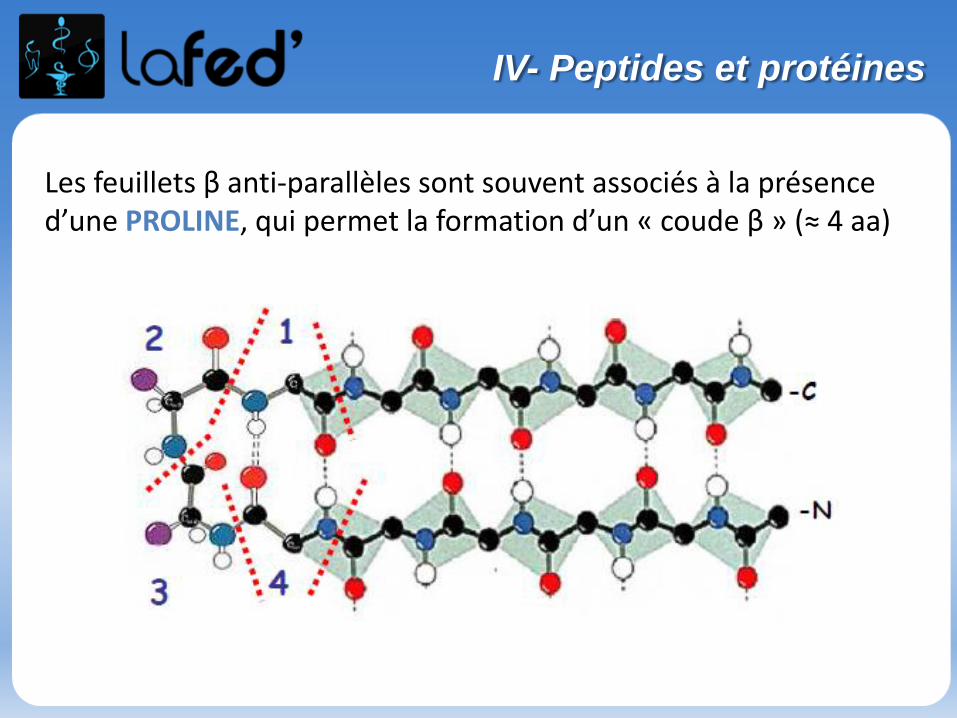
Les feuillets β anti-parallèles sont souvent associés à la présence d’une PROLINE, qui permet la formation d’un « coude β » (≈ 4 aa)
IV- Peptides et protéines
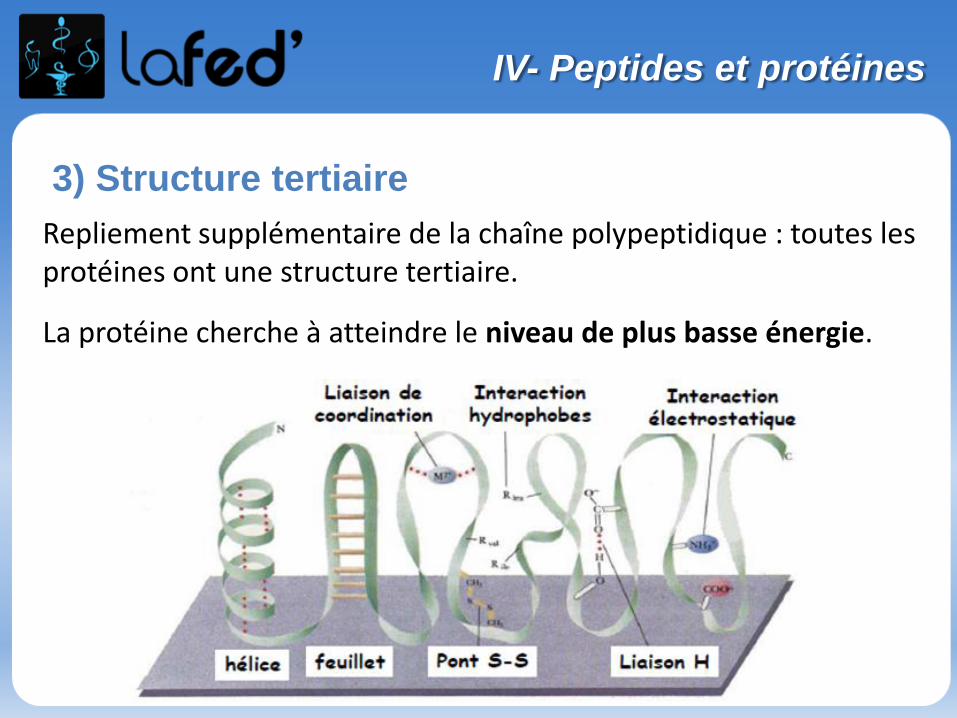
Repliement supplémentaire de la chaîne polypeptidique : toutes les protéines ont une structure tertiaire.
La protéine cherche à atteindre le niveau de plus basse énergie.
IV- Peptides et protéines
3) Structure tertiaire
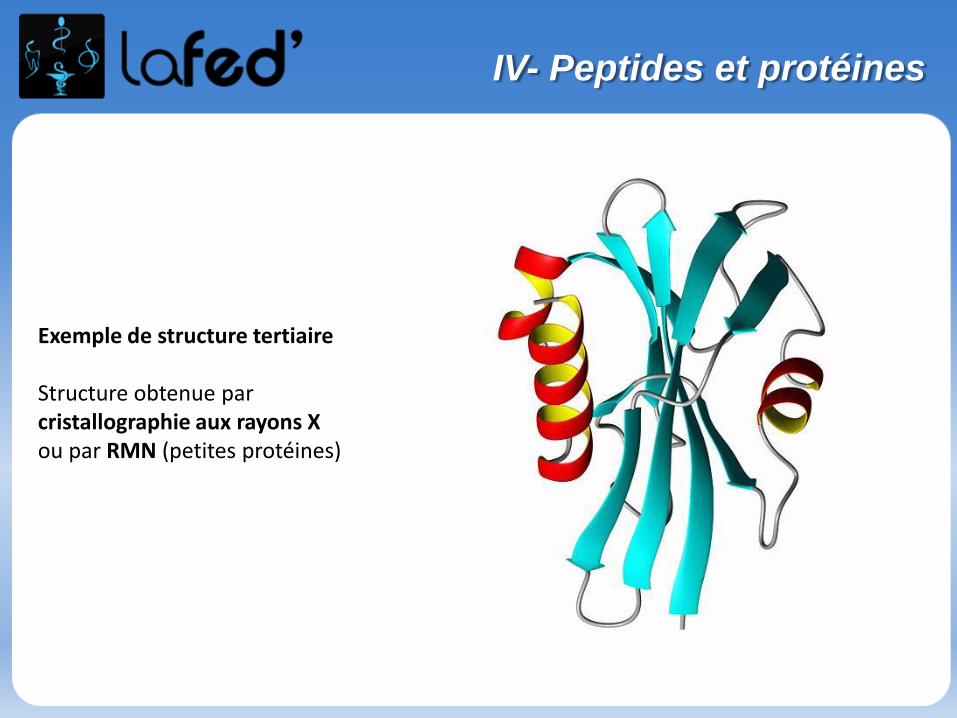
Exemple de structure tertiaire
Structure obtenue par cristallographie aux rayons X ou par RMN (petites protéines)
IV- Peptides et protéines

Toutes les protéines NE POSSEDENT PAS de structure quaternaire
La structure quaternaire correspond à l’association de plusieurs chaînes peptidiques par des liaisons faibles, comme des liaisons hydrogènes
IV- Peptides et protéines
4) Structure quaternaire
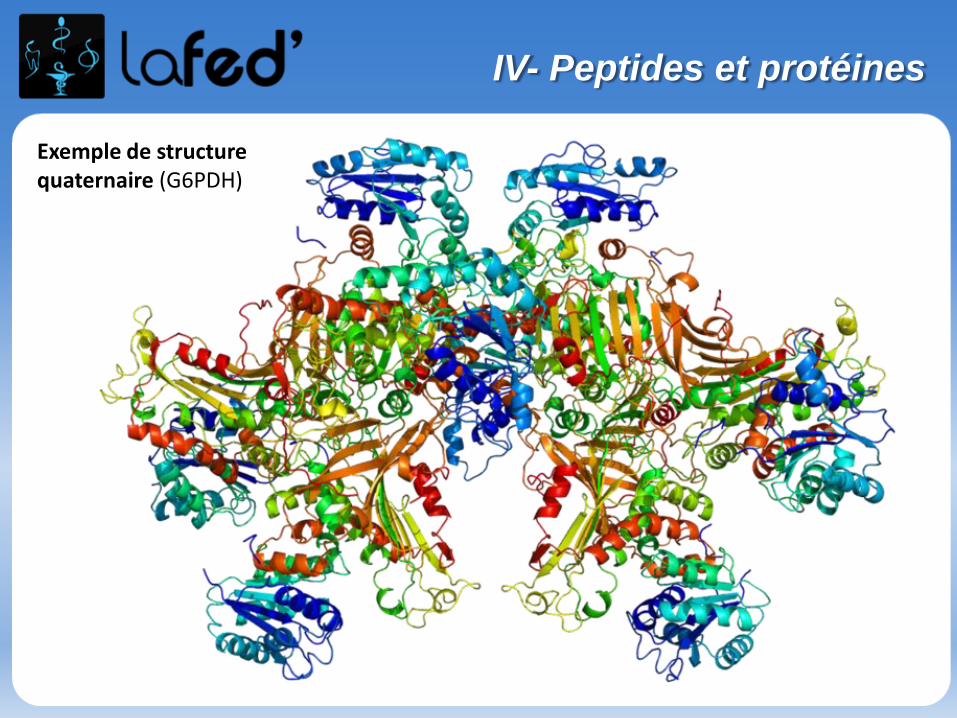
Exemple de structure quaternaire (G6PDH)
IV- Peptides et protéines

On peut dénaturer une protéine en « cassant » sa structure tertiaire
Elle passe de l’état « plié » a l’état « déplié » grâce à certains agents dénaturants comme :
- la chaleur
- pH extrêmes
- les détergents (SDS)
- …
IV- Peptides et protéines
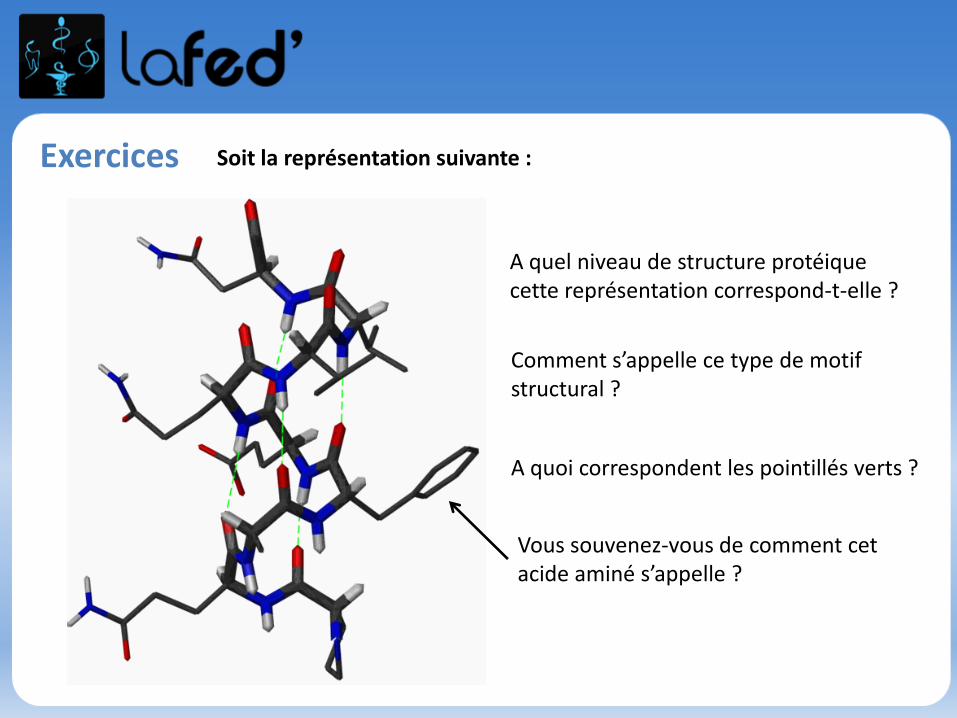
Exercices Soit la représentation suivante :
A quel niveau de structure protéiquecette représentation correspond-t-elle ?
Comment s’appelle ce type de motif structural ?
A quoi correspondent les pointillés verts ?
Vous souvenez-vous de comment cet acide aminé s’appelle ?
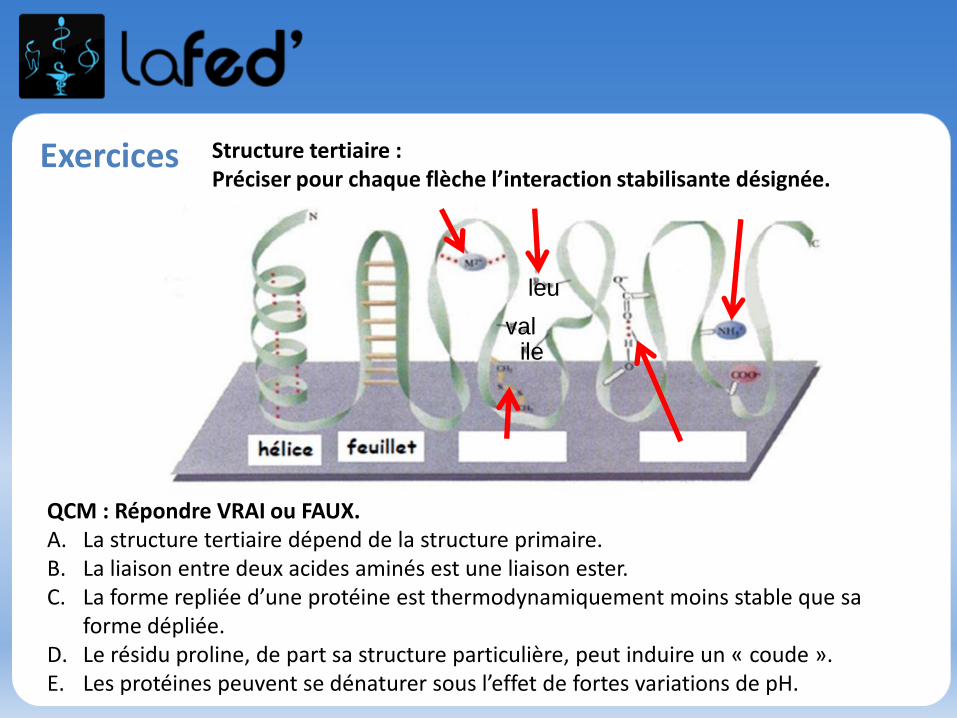
Exercices
QCM : Répondre VRAI ou FAUX.A. La structure tertiaire dépend de la structure primaire.B. La liaison entre deux acides aminés est une liaison ester.C. La forme repliée d’une protéine est thermodynamiquement moins stable que sa
forme dépliée.D. Le résidu proline, de part sa structure particulière, peut induire un « coude ».E. Les protéines peuvent se dénaturer sous l’effet de fortes variations de pH.
Structure tertiaire : Préciser pour chaque flèche l’interaction stabilisante désignée.
leu
valile

Exemple n°1 : l’hémoglobine
4 sous-unitésChez l’adulte : 2α + 2βChez le fœtus : 2α + 2γ
Rôle dans le transport de O2, protéine retrouvée dans les hématies.
Responsable de la coloration soit rouge, soit bleue du sang.
V- Exemples de protéines
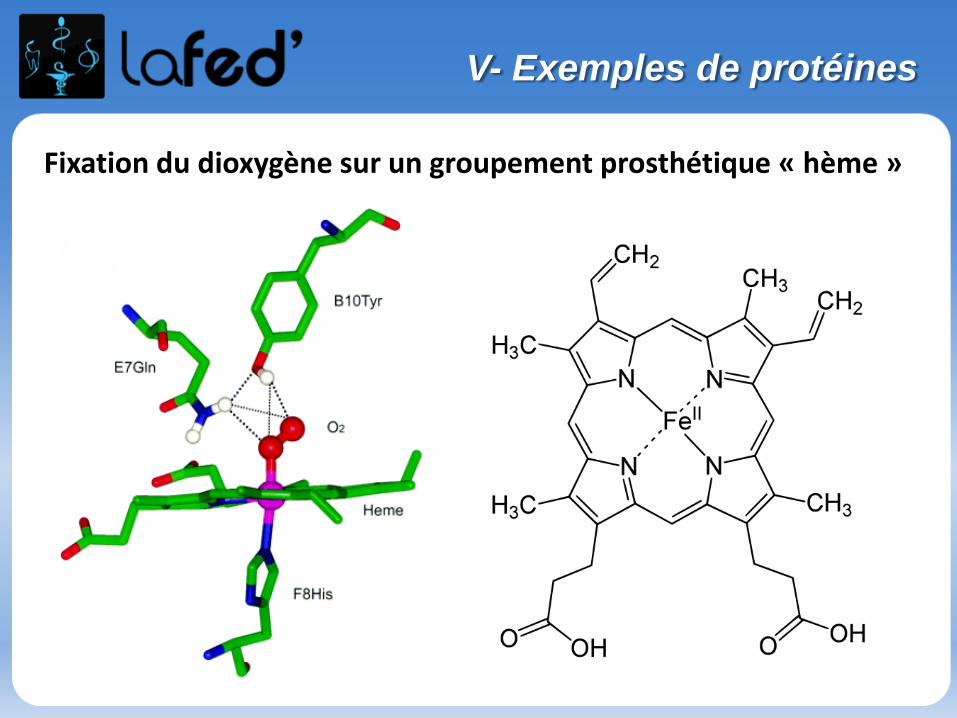
Fixation du dioxygène sur un groupement prosthétique « hème »
V- Exemples de protéines
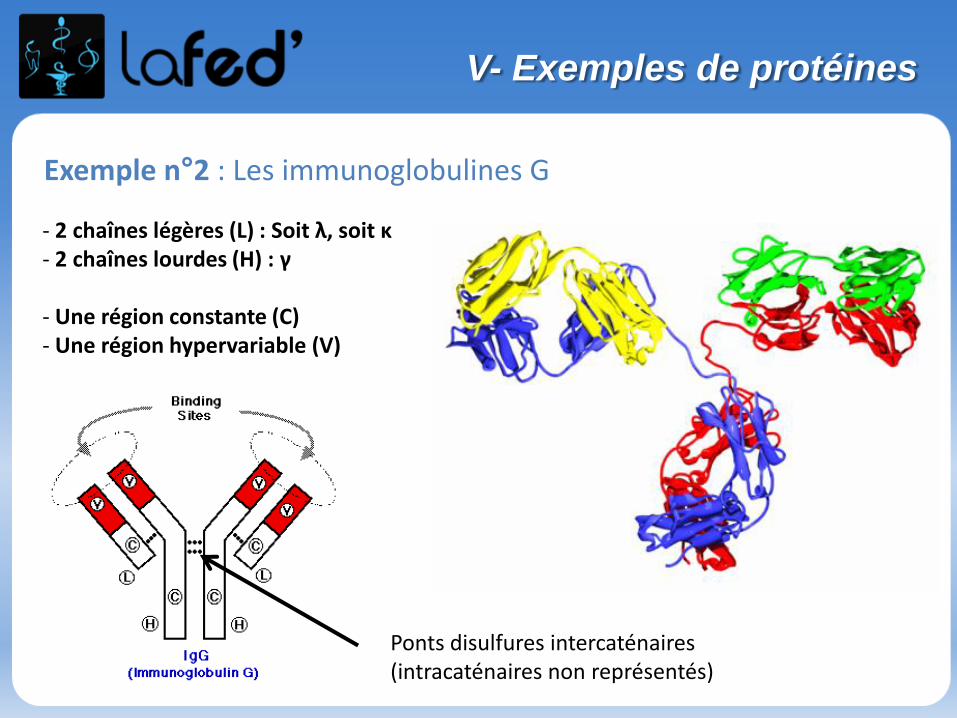
Exemple n°2 : Les immunoglobulines G
- 2 chaînes légères (L) : Soit λ, soit κ- 2 chaînes lourdes (H) : γ
- Une région constante (C)- Une région hypervariable (V)
Ponts disulfures intercaténaires(intracaténaires non représentés)
V- Exemples de protéines
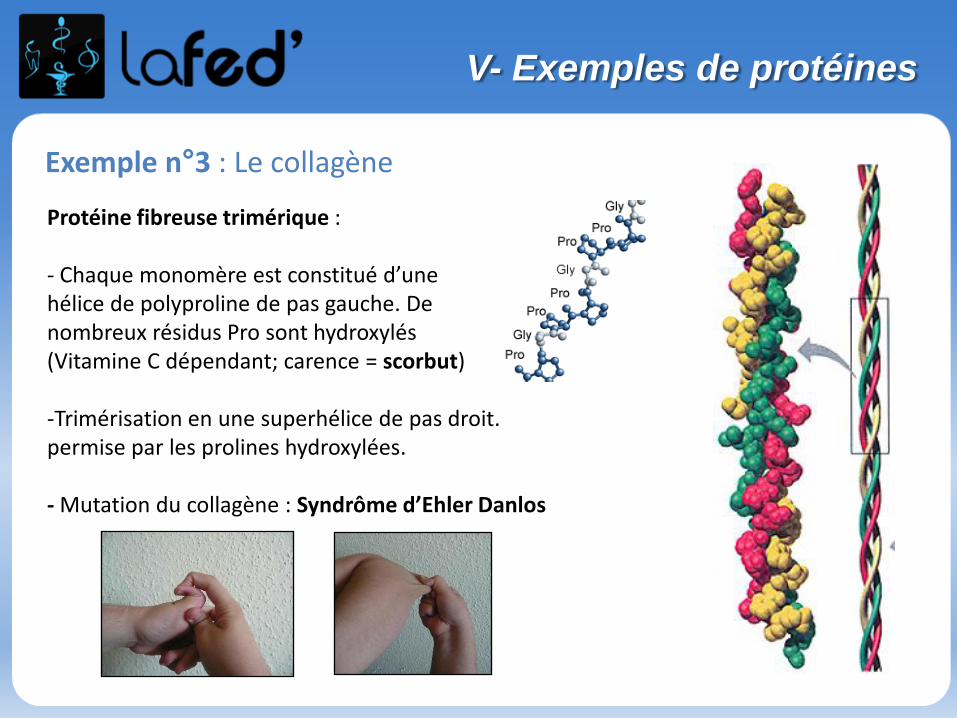
Protéine fibreuse trimérique :
- Chaque monomère est constitué d’unehélice de polyproline de pas gauche. Denombreux résidus Pro sont hydroxylés(Vitamine C dépendant; carence = scorbut)
-Trimérisation en une superhélice de pas droit.permise par les prolines hydroxylées.
- Mutation du collagène : Syndrôme d’Ehler Danlos
Exemple n°3 : Le collagène
V- Exemples de protéines

Exercices Questions pour un PACES !
1) Molécule aromatique de type tétrapyrole, jeforme un plan qui vient coordiner 4 fois un ionferreux Fe2+ le plus souvent. Je permets letransport du dioxygène, je suis, je suis, je suis… ?
2) Cofacteur de la proline hydroxylase, je permet labonne hydroxylation des prolines qui rentrent dansla constitution du collagène. Mon absenceprovoque le scorbut, je suis, je suis, je suis… ?
3) Acide aminé de formule brute C2H5O2N dans sa forme non ionisée, je rentre dansla composition de la gélatine et mon autre nom est la « glycocolle ». Je suis neutre àpH physiologique, acide aminé protéinogène le plus simple et constituant importantdu collagène et des coudes β, je suis, je suis, je suis… ?
V- Exemples de protéines

Stage de Pré-Rentrée 2012UE 1 – Les glucides
237
Diaporama réalisé par les tuteurs de La Fed’

• Les glucides sont les biomolécules les plusabondantes de l’organisme.
• Rôle dans le métabolisme énergétique +++Glucose = principal carburant cellulaire
• Rôle de structuration des édifices biologiquesCellulose = Polymère de structure des végétaux
• Autres rôles, parfois très divers…
I- Généralités
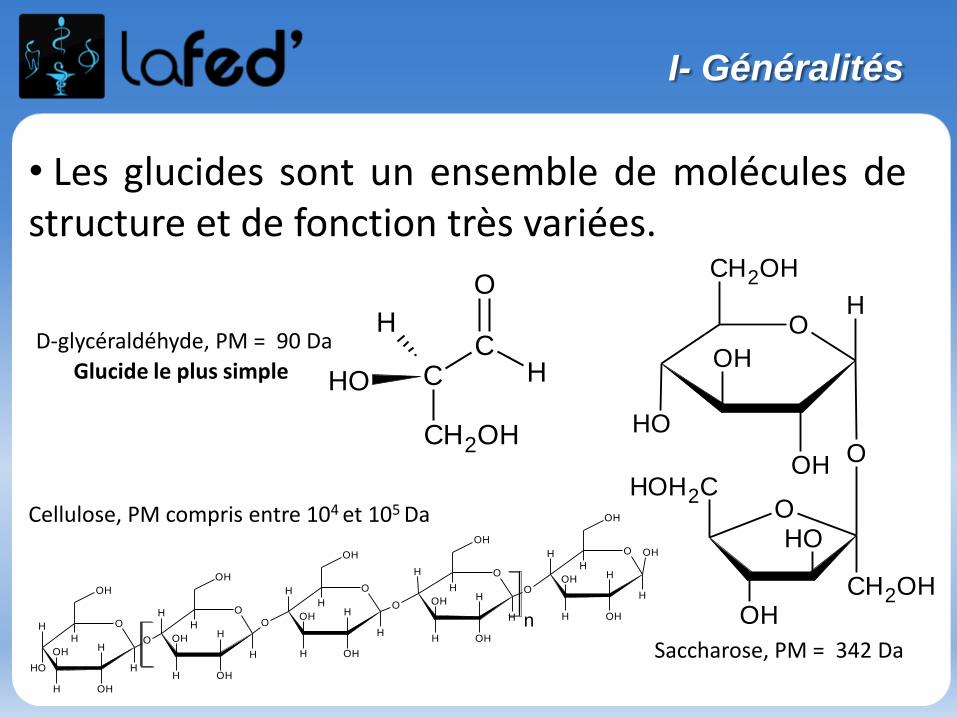
• Les glucides sont un ensemble de molécules destructure et de fonction très variées.
Saccharose, PM = 342 Da
OH
OOH
OH
OH
CH2OH
OOH
OHCH2OH
HOH2C
CH2OH
CC
H
OH
O
H
D-glycéraldéhyde, PM = 90 Da
O
H
HH
H
OOH
H OH
OH
O
H
HH
H
O OH
H OH
OH
O
H
HH
H
OOH
H OH
OH
O
H
HH
H
O OH
H OH
OH
O
H
HH
H
OH
OH
H OH
OH
OH
n
Cellulose, PM compris entre 104 et 105 Da
Glucide le plus simple
I- Généralités

• Il est néanmoins possible de dégager unecaractéristique commune.
• Glucide = Toute molécule contenant au moins unose (ou saccharide) ou dérivé d’ose.
• Grossièrement on peut dire que: Les oses sont auxglucides ce que les acides aminés sont auxprotéines (des briques élémentaires).
I- Généralités
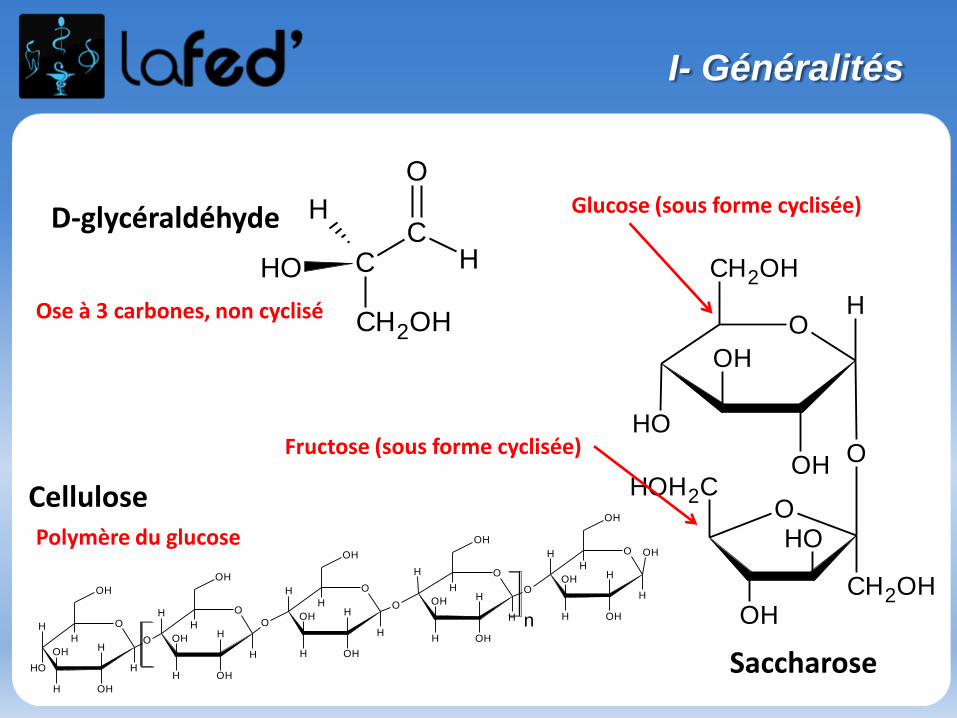
Saccharose
OH
OOH
OH
OH
CH2OH
OOH
OHCH2OH
HOH2C
CH2OH
CC
H
OH
O
H
D-glycéraldéhyde
Cellulose
Ose à 3 carbones, non cyclisé
Glucose (sous forme cyclisée)
Fructose (sous forme cyclisée)
Polymère du glucoseO
H
HH
H
OOH
H OH
OH
O
H
HH
H
O OH
H OH
OH
O
H
HH
H
OOH
H OH
OH
O
H
HH
H
O OH
H OH
OH
O
H
HH
H
OH
OH
H OH
OH
OH
n
I- Généralités

• Molécules simples non hydrolysables, parfoisappelés hydrates de carbone.
• Formule générale:
Un équivalent H2O pour chaque atome de carbone.
Cn(H2O)n n ≥ 3
I- Généralités

• Si n = 3 : triose (C3H6O3) ex : glycéraldéhyde, DHA
• Si n = 4 : tétrose (C4H8O4) ex : érythrose, thréose
• Si n = 5 : pentose (C5H10O5) ex : ribose
• Si n = 6 : hexose (C6H12O6) ex : glucose, fructose
• Si n = 7 : heptose (C7 H14O7) ex : sédoheptuloseseul exemple chez l’homme
Pas d’octose (n = 8) chez le mammifère.
• Formule générale:Un équivalent H2O pour chaque atome de carbone.
I- Généralités
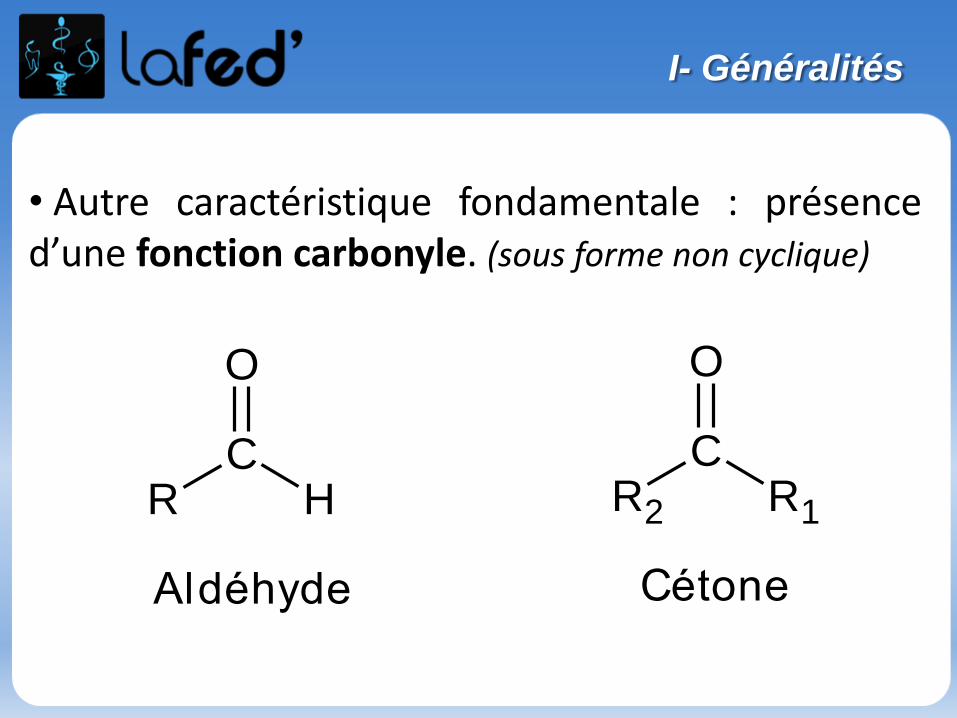
• Autre caractéristique fondamentale : présenced’une fonction carbonyle. (sous forme non cyclique)
C
O
R2 R1
Cétone
C
O
R H
Aldéhyde
I- Généralités
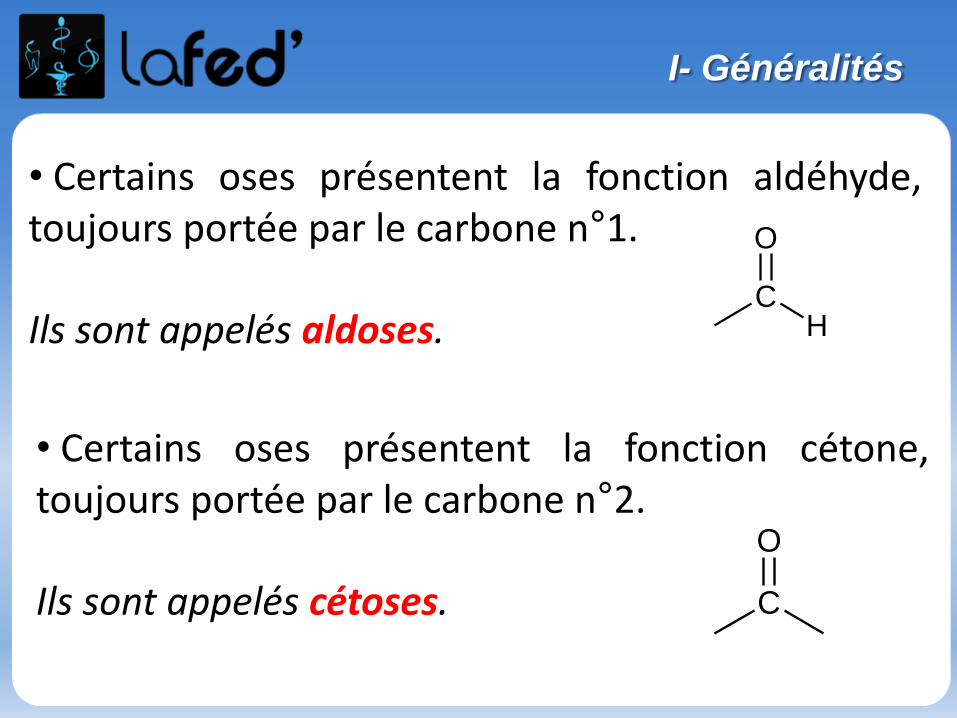
• Certains oses présentent la fonction aldéhyde,toujours portée par le carbone n°1.
Ils sont appelés aldoses.C
H
O
• Certains oses présentent la fonction cétone,toujours portée par le carbone n°2.
Ils sont appelés cétoses. C
O
I- Généralités
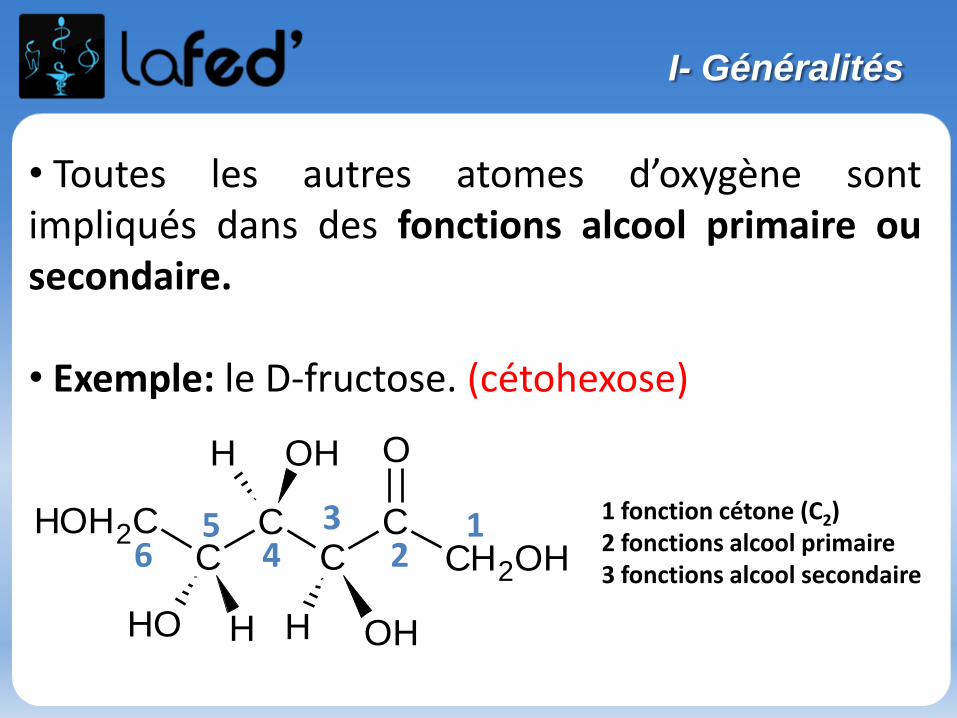
• Toutes les autres atomes d’oxygène sontimpliqués dans des fonctions alcool primaire ousecondaire.
• Exemple: le D-fructose. (cétohexose)
CC
CC
HOH2CCH2OH
OH
OH
OH
OH
H H
1 fonction cétone (C2)2 fonctions alcool primaire3 fonctions alcool secondaire
213
45
6
I- Généralités
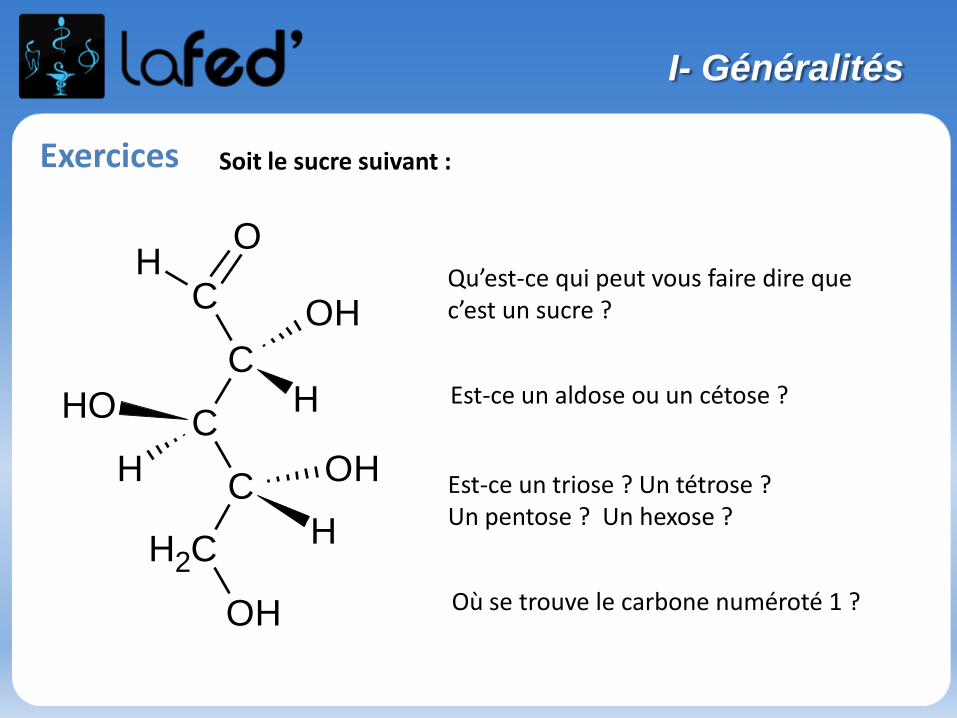
Exercices
C
C
C
C
CH2
O
OH
OH
OH
OH
H
H
H
H
Soit le sucre suivant :
Qu’est-ce qui peut vous faire dire que c’est un sucre ?
Est-ce un aldose ou un cétose ?
Est-ce un triose ? Un tétrose ? Un pentose ? Un hexose ?
Où se trouve le carbone numéroté 1 ?
I- Généralités
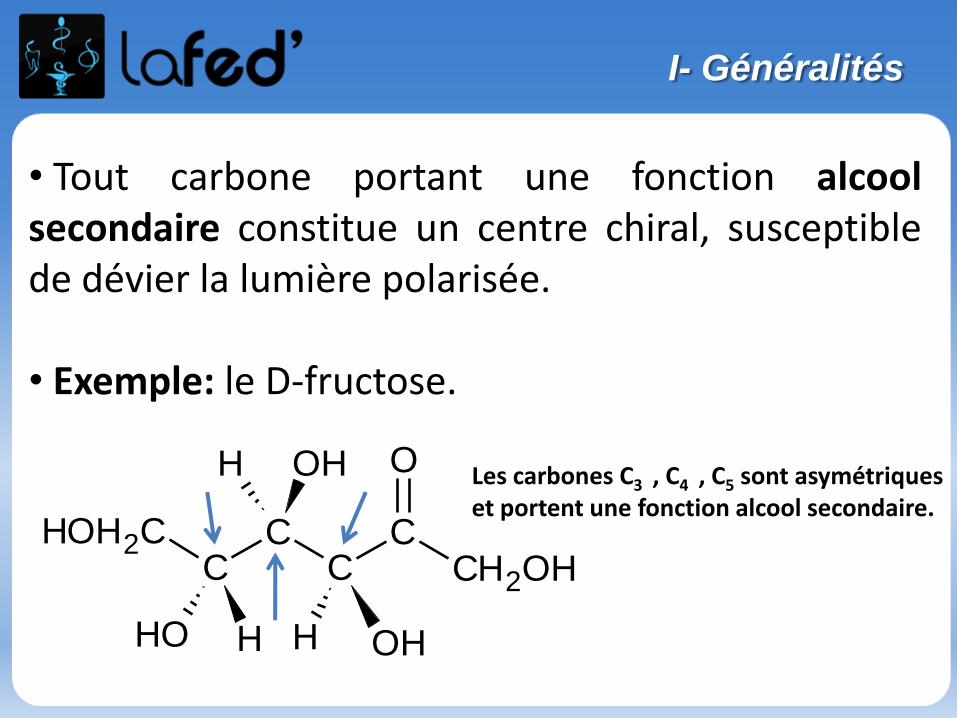
• Tout carbone portant une fonction alcoolsecondaire constitue un centre chiral, susceptiblede dévier la lumière polarisée.
• Exemple: le D-fructose.
CC
CC
HOH2CCH2OH
OH
OH
OH
OH
H H
Les carbones C3 , C4 , C5 sont asymétriqueset portent une fonction alcool secondaire.
I- Généralités
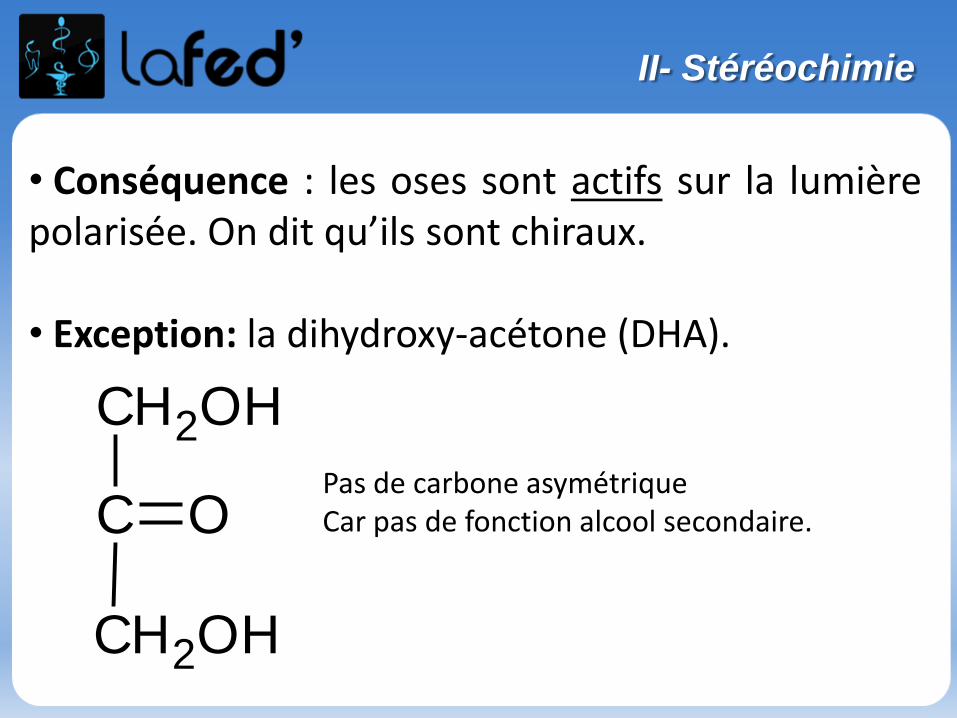
• Conséquence : les oses sont actifs sur la lumièrepolarisée. On dit qu’ils sont chiraux.
• Exception: la dihydroxy-acétone (DHA).
CH2OH
C
CH2OH
OPas de carbone asymétriqueCar pas de fonction alcool secondaire.
II- Stéréochimie
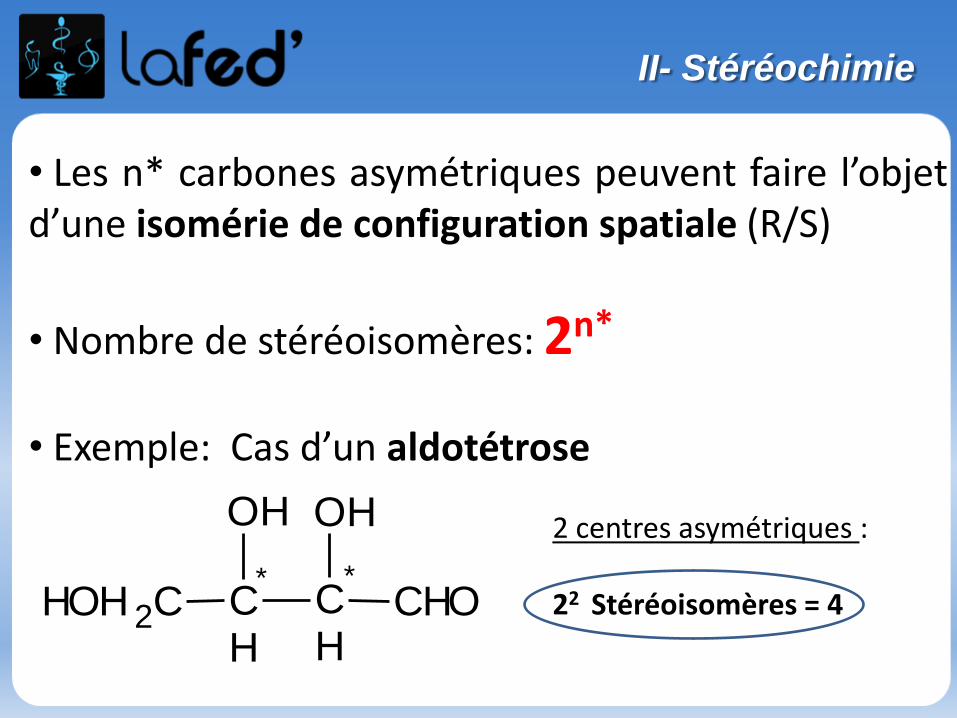
• Les n* carbones asymétriques peuvent faire l’objetd’une isomérie de configuration spatiale (R/S)
• Nombre de stéréoisomères: 2n*
• Exemple: Cas d’un aldotétrose
CHOCH
CH
HOH 2C
OHOH
* *
2 centres asymétriques :
22 Stéréoisomères = 4
II- Stéréochimie
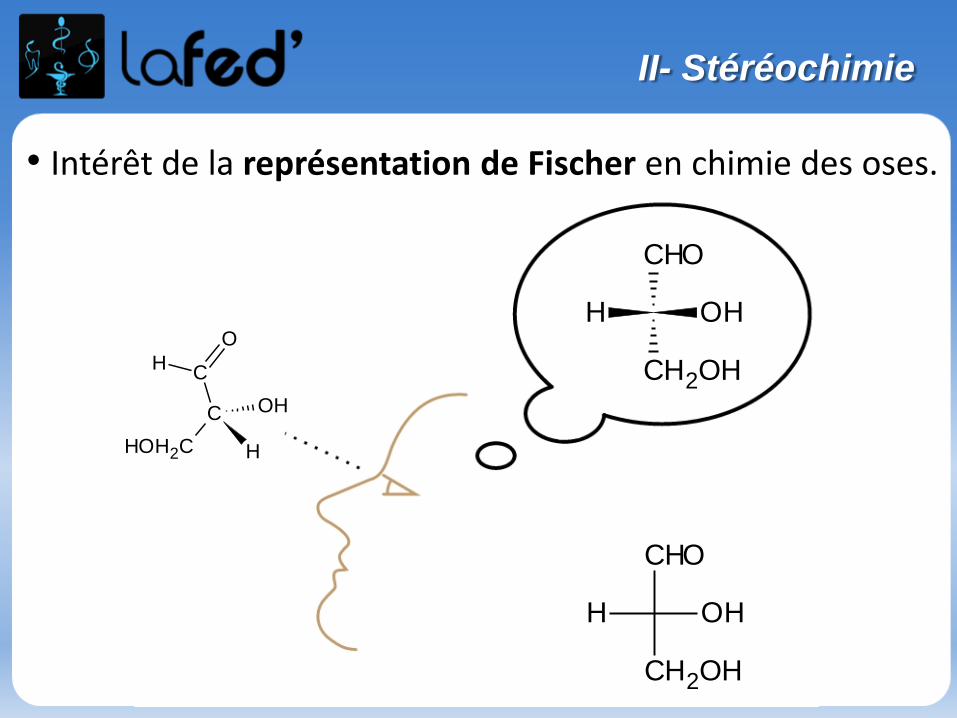
• Intérêt de la représentation de Fischer en chimie des oses.
HOH2C
C
C
OH
H
O
H
CHO
CH2OH
H OH
CHO
CH2OH
H OH
II- Stéréochimie
• Cas de la molécule de glycéraldéhyde.(21 = 2 stéréoisomères)
CHO
CH2OH
H OH
CHO
CH2OH
OH H
D-glycéraldéhyde
[] = +14°
L-glycéraldéhyde
[] = -14°
différent de
II- Stéréochimie

• Il existe deux séries d’oses : la série D et la série L.
• Les oses de la série D sont énantiomères des oses de lasérie L.
• La série D est la plus représentée dans la nature.
• Détermination de la série d’un ose :Projection de Fischer +++
II- Stéréochimie
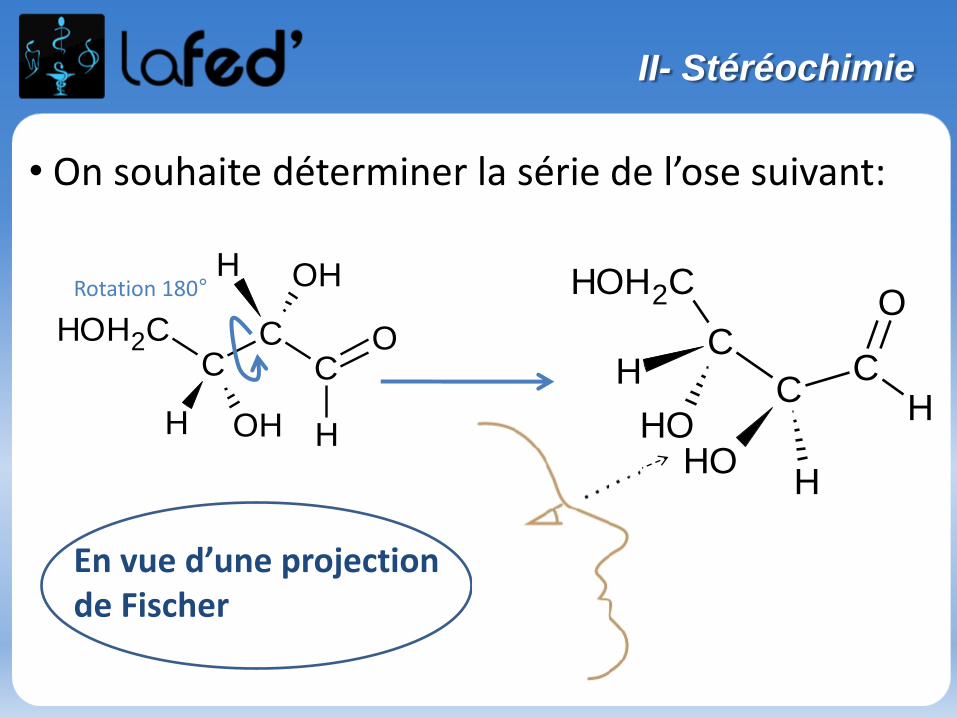
• On souhaite déterminer la série de l’ose suivant:
CC
CHOH2C O
OH
OH HH
H
En vue d’une projection de Fischer
Rotation 180°
CC
C
HOH2CO
OHOH
HH
H
II- Stéréochimie
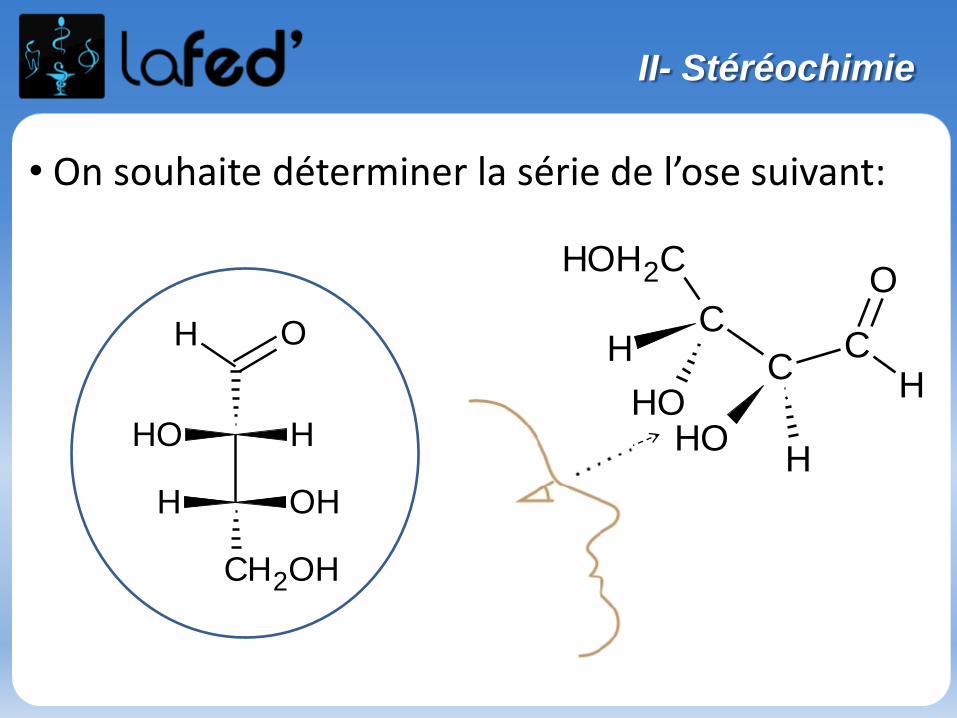
• On souhaite déterminer la série de l’ose suivant:
CC
C
HOH2CO
OHOH
HH
H
CH2OH
O
OH H
H OH
H
II- Stéréochimie
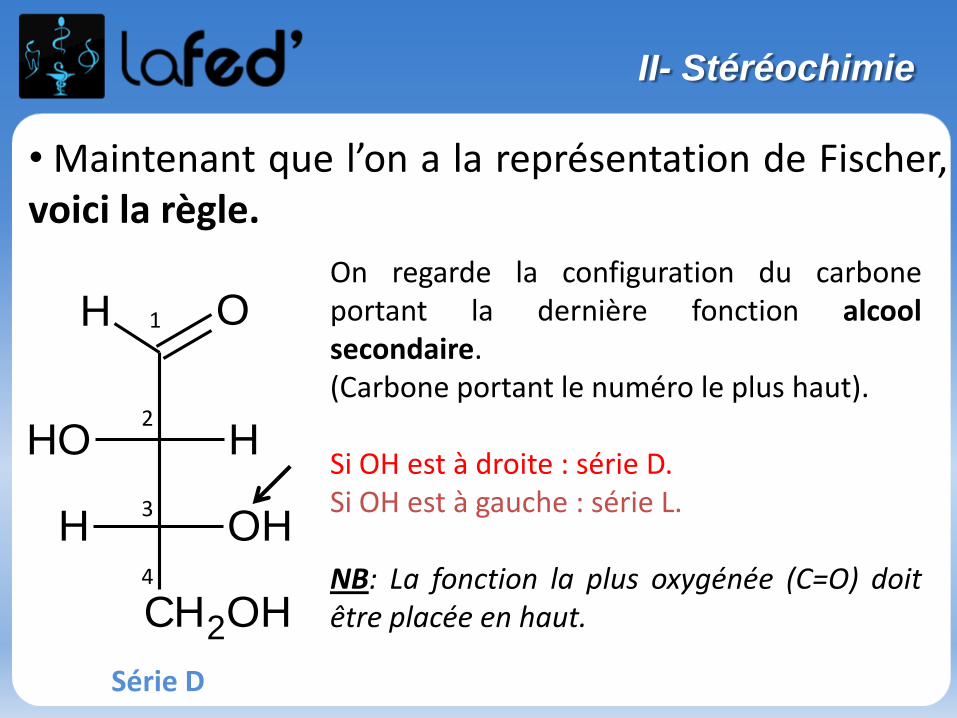
• Maintenant que l’on a la représentation de Fischer,voici la règle.
CH2OH
O
OH H
H OH
HOn regarde la configuration du carboneportant la dernière fonction alcoolsecondaire.(Carbone portant le numéro le plus haut).
Si OH est à droite : série D.Si OH est à gauche : série L.
NB: La fonction la plus oxygénée (C=O) doitêtre placée en haut.
1
2
3
4
Série D
II- Stéréochimie
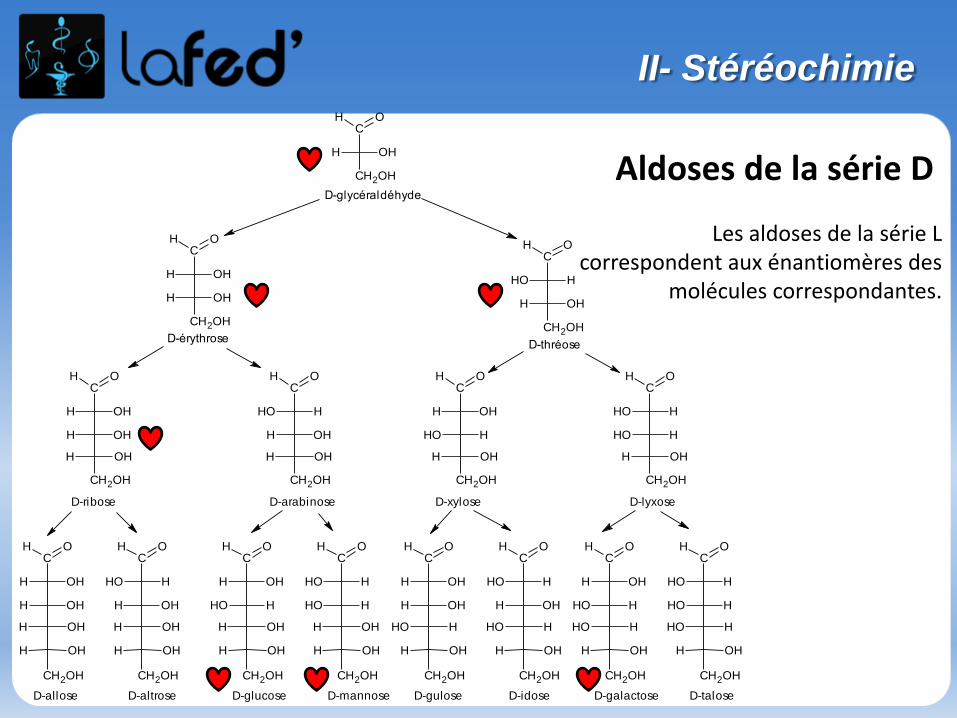
Aldoses de la série D
Les aldoses de la série L correspondent aux énantiomères des
molécules correspondantes.
C
CH2OH
O
H OH
H OH
H
C
CH2OH
O
H OH
H
C
CH2OH
O
OH H
H OH
H
D-glycéraldéhyde
D-érythroseD-thréose
CO
H OH
H OH
H
CH2OH
OHH
D-ribose
CO
OH H
H OH
H
CH2OH
OHH
D-arabinose
CO
H OH
OH H
H
CH2OH
OHH
D-xylose
CO
OH H
OH H
H
CH2OH
OHH
D-lyxose
CO
H OH
H OH
H
OHH
CH2OH
OHH
D-allose
CO
OH H
H OH
H
OHH
CH2OH
OHH
D-altrose
CO
H OH
OH H
H
OHH
CH2OH
OHH
D-glucose
CO
OH H
OH H
H
OHH
CH2OH
OHH
D-mannose
CO
H OH
H OH
H
HOH
CH2OH
OHH
D-gulose
CO
OH H
H OH
H
HOH
CH2OH
OHH
D-idose
CO
H OH
OH H
H
HOH
CH2OH
OHH
D-galactose
CO
OH H
OH H
H
HOH
CH2OH
OHH
D-talose
II- Stéréochimie
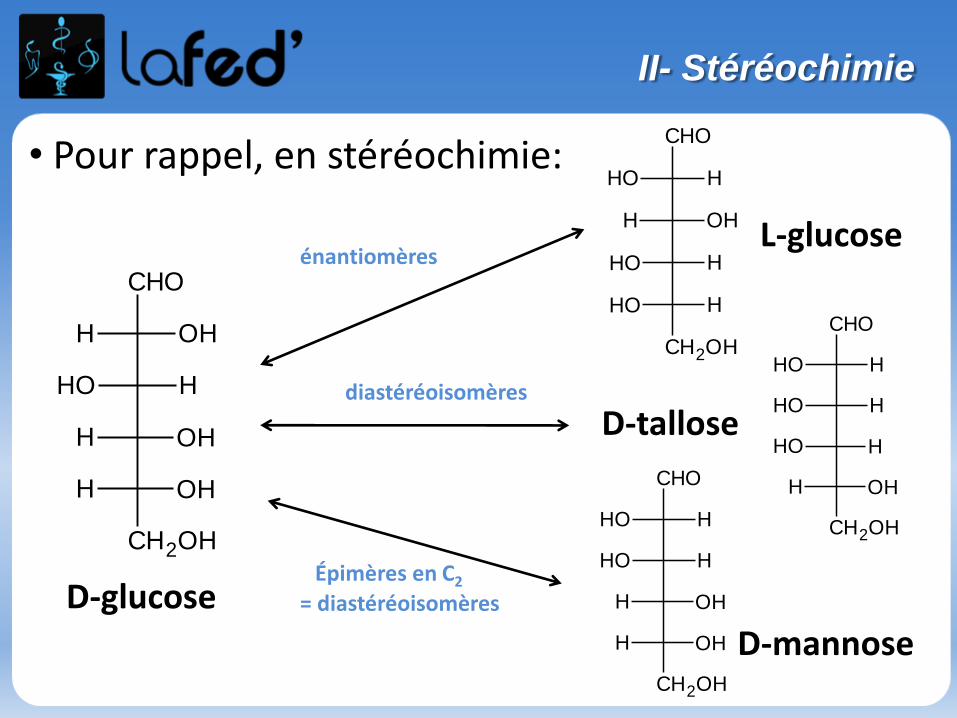
• Pour rappel, en stéréochimie:
CHO
H OH
OH H
OHH
OH
CH2OH
H
D-glucose
CHO
HOH
OHH
OH H
OH
CH2OH
H
L-glucose
CHO
OH H
OH H
OHH
OH
CH2OH
H D-mannose
CHO
OH H
OH H
HOH
OH
CH2OH
H
D-tallose
énantiomères
diastéréoisomères
Épimères en C2
= diastéréoisomères
II- Stéréochimie
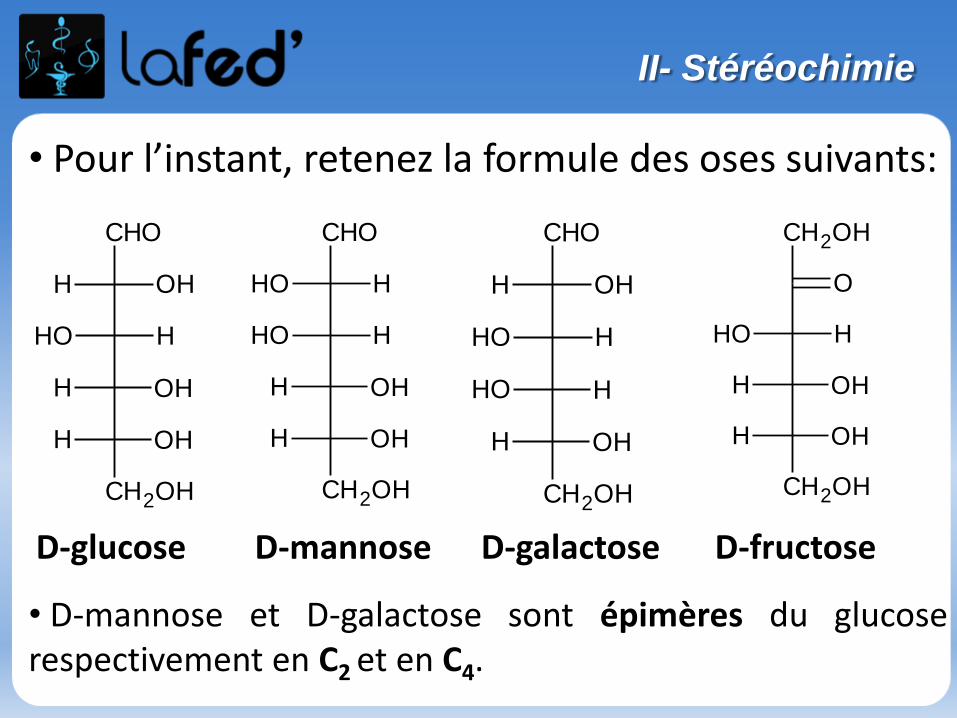
• Pour l’instant, retenez la formule des oses suivants:
• D-mannose et D-galactose sont épimères du glucoserespectivement en C2 et en C4.
CHO
H OH
OH H
OHH
OH
CH2OH
H
D-glucose
CHO
OH H
OH H
OHH
OH
CH2OH
H
D-mannose
CHO
H OH
OH H
HOH
OH
CH2OH
H
D-galactose D-fructose
CH2OH
O
OH H
OHH
OH
CH2OH
H
II- Stéréochimie
• Autres oses d’intérêt biologique à retenir :
CHO
H OH
OHH
OH
CH2OH
H
D-ribose (constituant des acides
nucléiques : ARN)
CHO
CH2OH
H OH
CH2OH
CH2OH
O
D-glycéraldéhyde
Dihydroxyacétone
Métabolites importants
II- Stéréochimie
CHO
H OH
OH H
OHH
OH
CH2OH
H
CHO
OH H
OH H
OHH
OH
CH2OH
H
Exercices Soient les deux sucres suivants :
Quelle est la série (D/L) de ces deux oses ?Quelle est la relation stéréochimique qui existent entre eux ?Comment s’appellent-ils ?
II- Stéréochimie
• En milieu aqueux, les oses sont retrouvés trèsmajoritairement sous forme cyclique.
OOH
OH
OH
OH
CH2OH
H
β-D-glucopyranoseUne des formes cycliques du D-glucose.
III- Cyclisation
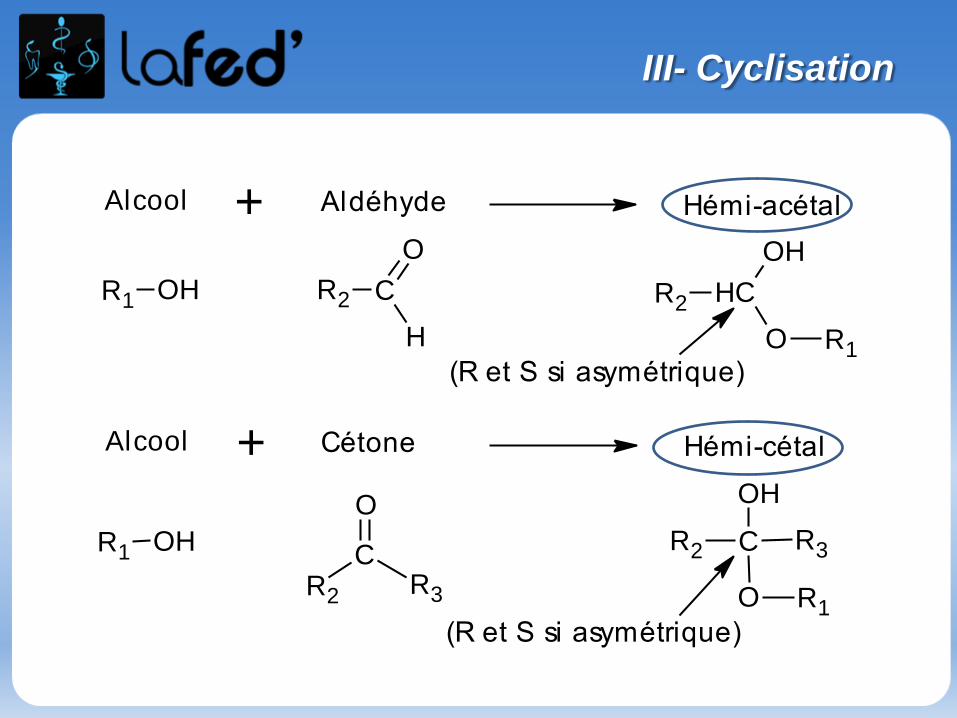
Alcool + Aldéhyde Hémi-acétal
Alcool + Cétone Hémi-cétal
R2
CR3
O
R2 C
H
O
R1 OH R2 CH
O
OH
R1(R et S si asymétrique)
R2 C
O
OH
R1
R3
(R et S si asymétrique)
R1 OH
III- Cyclisation
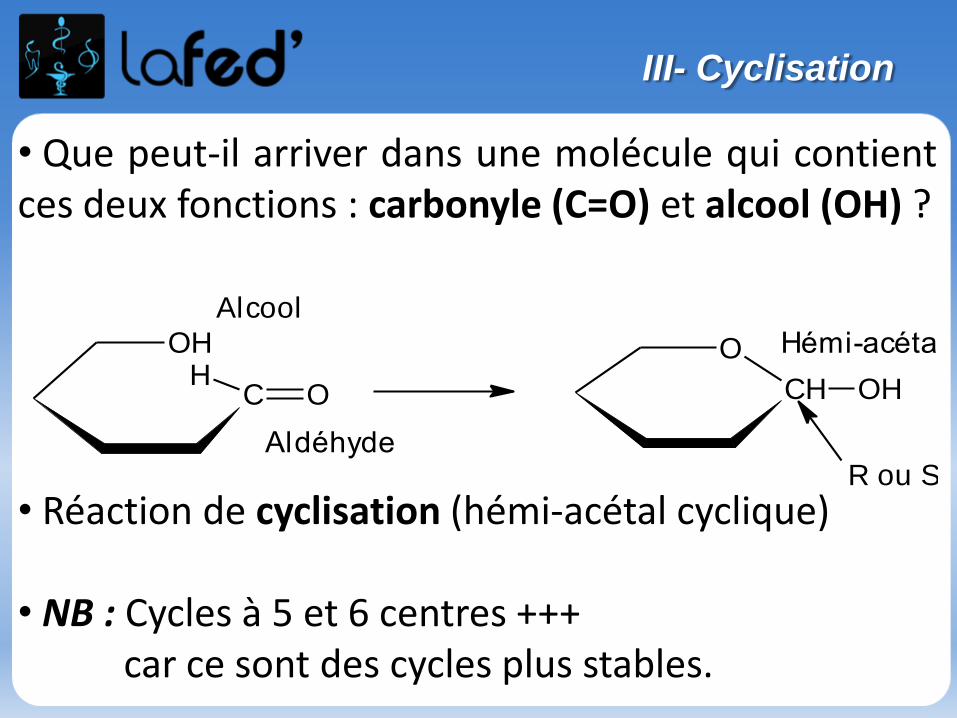
• Que peut-il arriver dans une molécule qui contientces deux fonctions : carbonyle (C=O) et alcool (OH) ?
• Réaction de cyclisation (hémi-acétal cyclique)
• NB : Cycles à 5 et 6 centres +++car ce sont des cycles plus stables.
OH
C OH
O
CH OH
Alcool
Aldéhyde
Hémi-acétal
R ou S
III- Cyclisation
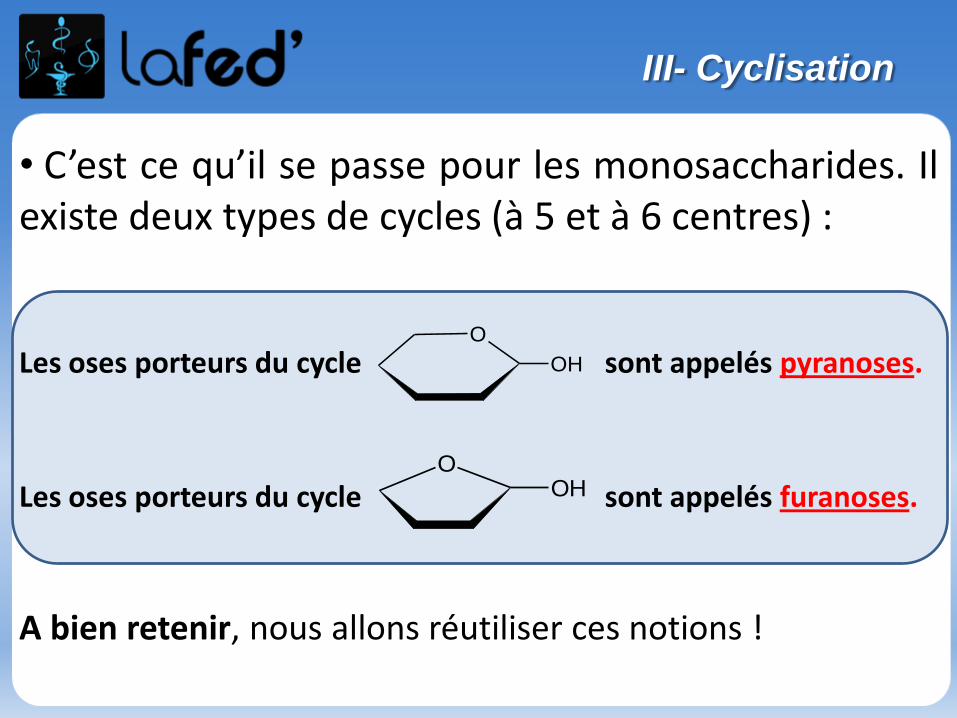
• C’est ce qu’il se passe pour les monosaccharides. Ilexiste deux types de cycles (à 5 et à 6 centres) :
Les oses porteurs du cycle sont appelés pyranoses.
Les oses porteurs du cycle sont appelés furanoses.
A bien retenir, nous allons réutiliser ces notions !
O
OH
OOH
III- Cyclisation
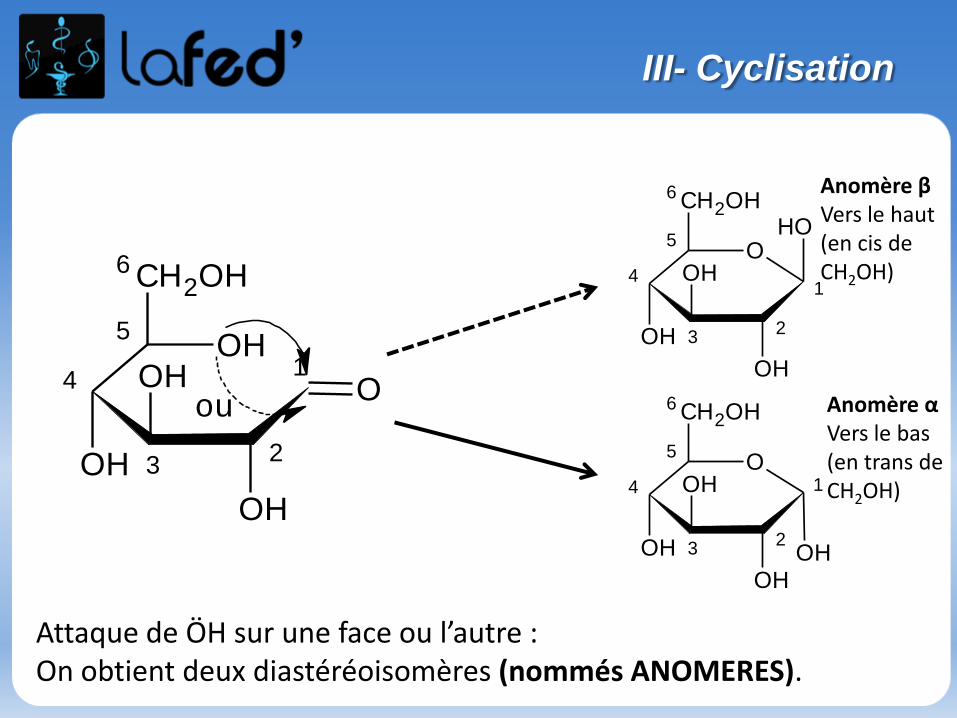
1O
OH
OH
OH
CH2OH
OH
6
5
4
32
ou
Attaque de ÖH sur une face ou l’autre : On obtient deux diastéréoisomères (nommés ANOMERES).
1
OH
OH
OH
OH
CH2OH
O
6
5
4
32
1
OHOH
OH
OH
CH2OH
O
6
5
4
32
Anomère βVers le haut(en cis de CH2OH)
Anomère αVers le bas(en trans de CH2OH)
III- Cyclisation
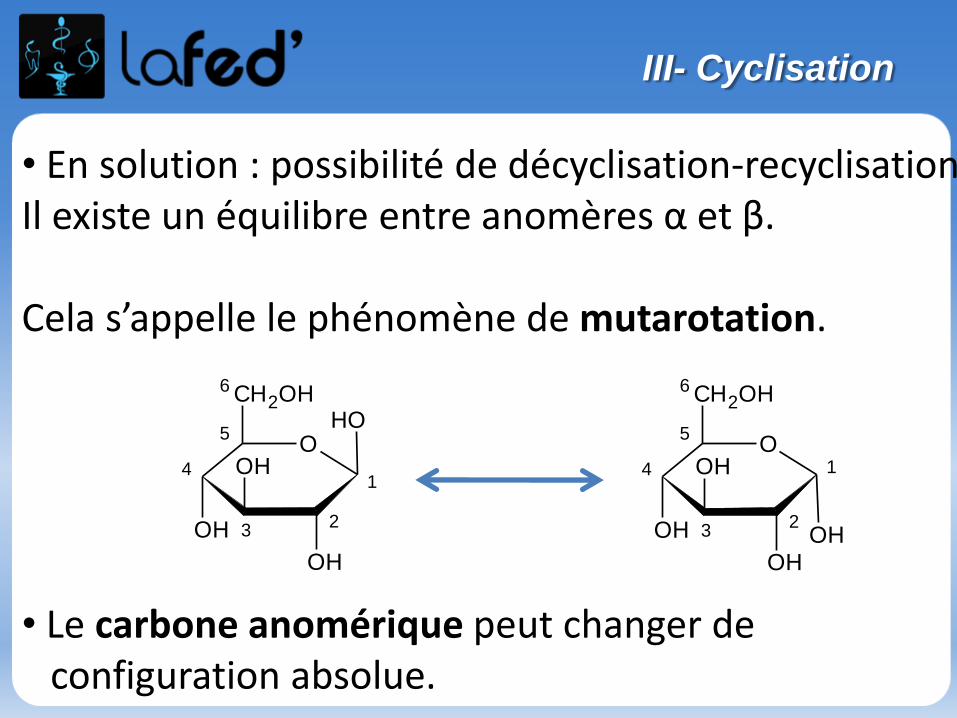
• En solution : possibilité de décyclisation-recyclisation.Il existe un équilibre entre anomères α et β.
Cela s’appelle le phénomène de mutarotation.
• Le carbone anomérique peut changer deconfiguration absolue.
1
OH
OH
OH
OH
CH2OH
O
6
5
4
32
1
OHOH
OH
OH
CH2OH
O
6
5
4
32
III- Cyclisation
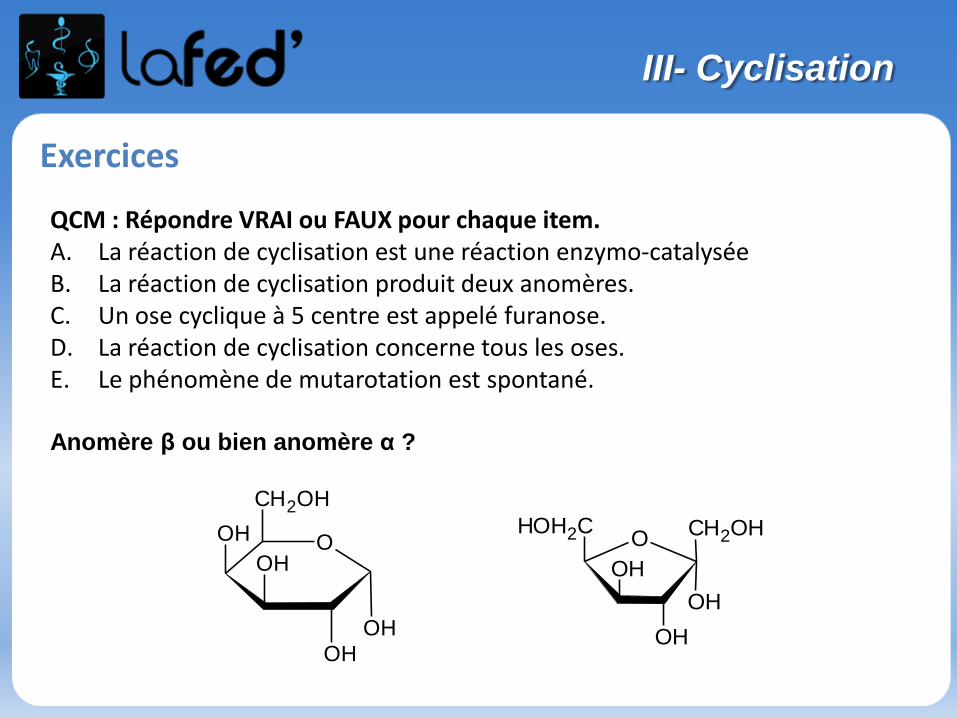
III- Cyclisation
Exercices
QCM : Répondre VRAI ou FAUX pour chaque item.A. La réaction de cyclisation est une réaction enzymo-catalyséeB. La réaction de cyclisation produit deux anomères.C. Un ose cyclique à 5 centre est appelé furanose.D. La réaction de cyclisation concerne tous les oses.E. Le phénomène de mutarotation est spontané.
Anomère β ou bien anomère α ?
OHOH
OH
OH
CH2OH
O O
OH
OH
HOH2C
OH
CH2OH
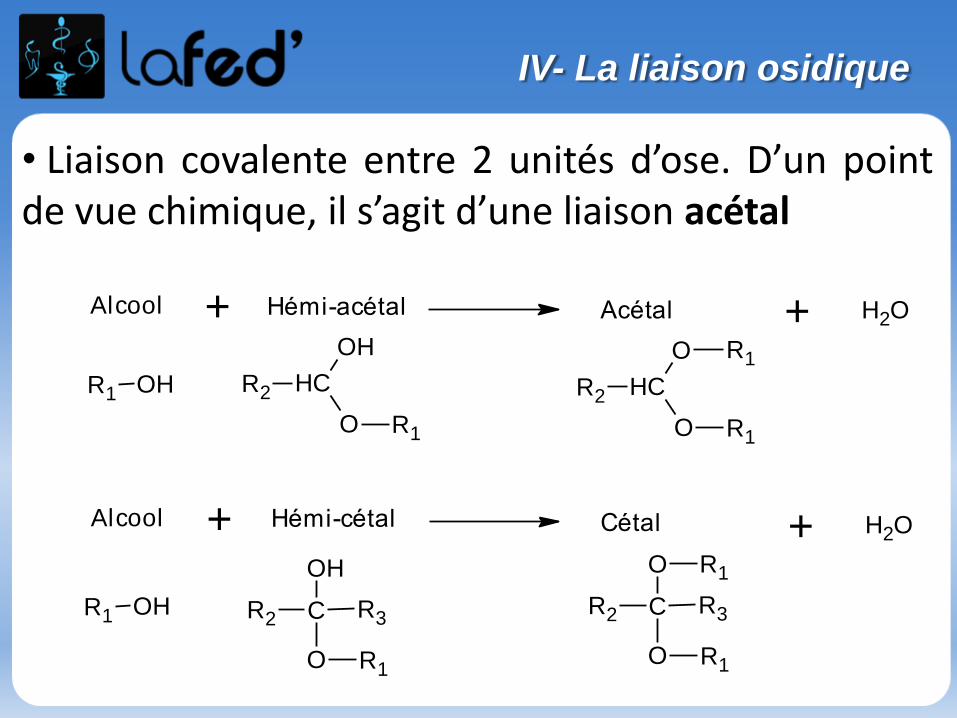
• Liaison covalente entre 2 unités d’ose. D’un pointde vue chimique, il s’agit d’une liaison acétal
Alcool + Acétal
Alcool + Cétal
R1 OH
R2 C
O
O
R1
R3
R1
R1 OH
Hémi-acétal
R2 CH
O
OH
R1
R2 CH
O
O
R1
R1
R2 C
O
OH
R1
R3
Hémi-cétal
+ OH2
+ OH2
IV- La liaison osidique
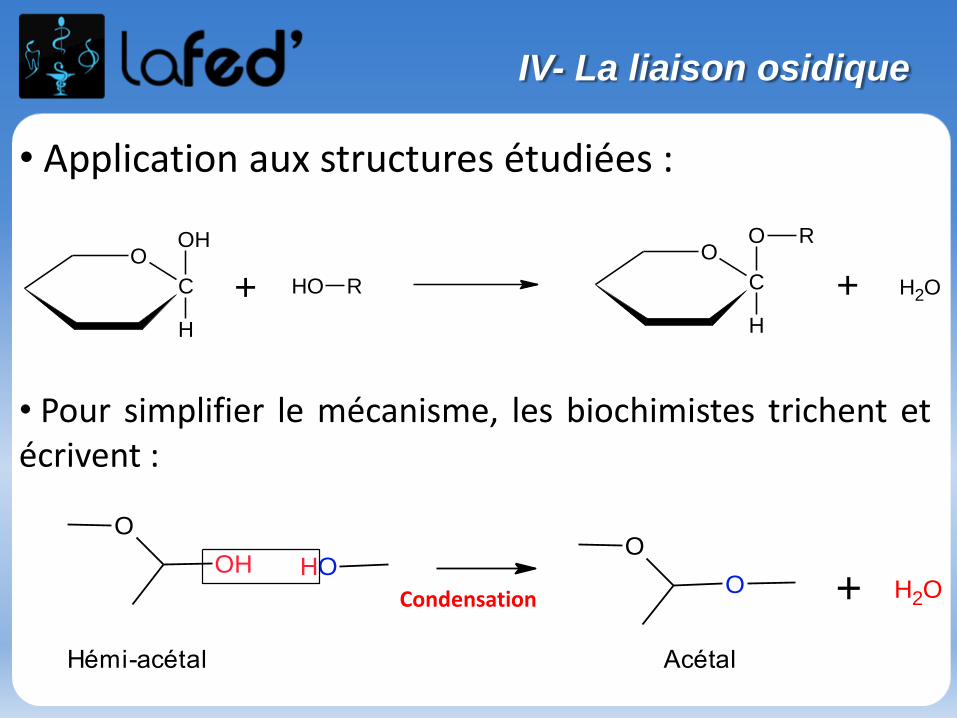
• Application aux structures étudiées :
• Pour simplifier le mécanisme, les biochimistes trichent etécrivent :
O
C
OH
H
OH R+O
C
O
H
R
+ OH2
OHOH
O
O
O
+ OH2
Hémi-acétal Acétal
Condensation
IV- La liaison osidique

• Le groupement OH, réagissant avec l’hémi-acétal peut :- être un alcool primaire- être un alcool secondaire- appartenir à une autre fonction hémi-acétal
• Si une liaison hémi-acétal est impliquée dans une liaisonosidique, la mutarotation de l’ose correspondant estbloquée.
• On définit alors des ponts osidiques α ou β en fonction del’anomérie du carbone hémi-acétalique bloqué.
IV- La liaison osidique
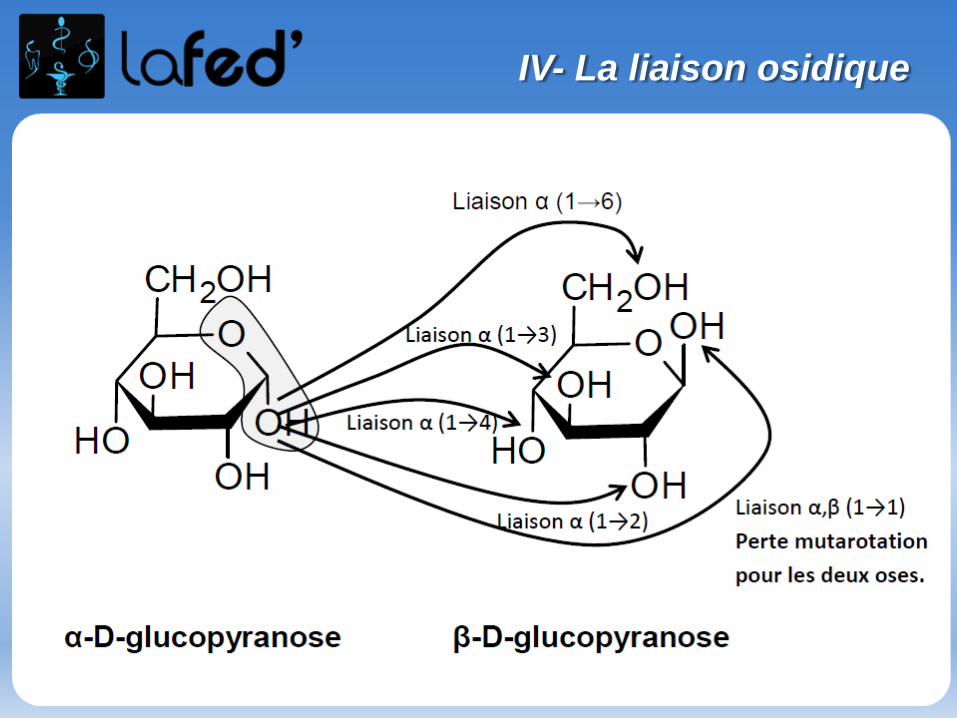
IV- La liaison osidique
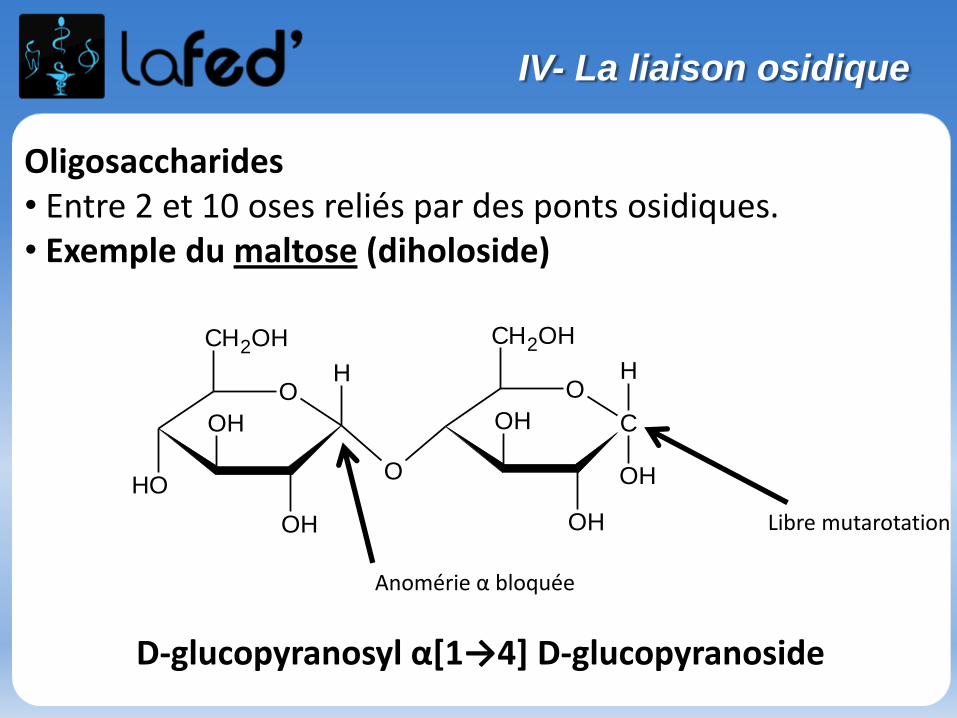
Oligosaccharides• Entre 2 et 10 oses reliés par des ponts osidiques.• Exemple du maltose (diholoside)
D-glucopyranosyl α*1→4+ D-glucopyranoside
OH
O
OH
OH
OH
CH2OH
O
C
H
OH
OH
CH2OH
OH
Libre mutarotation
Anomérie α bloquée
IV- La liaison osidique
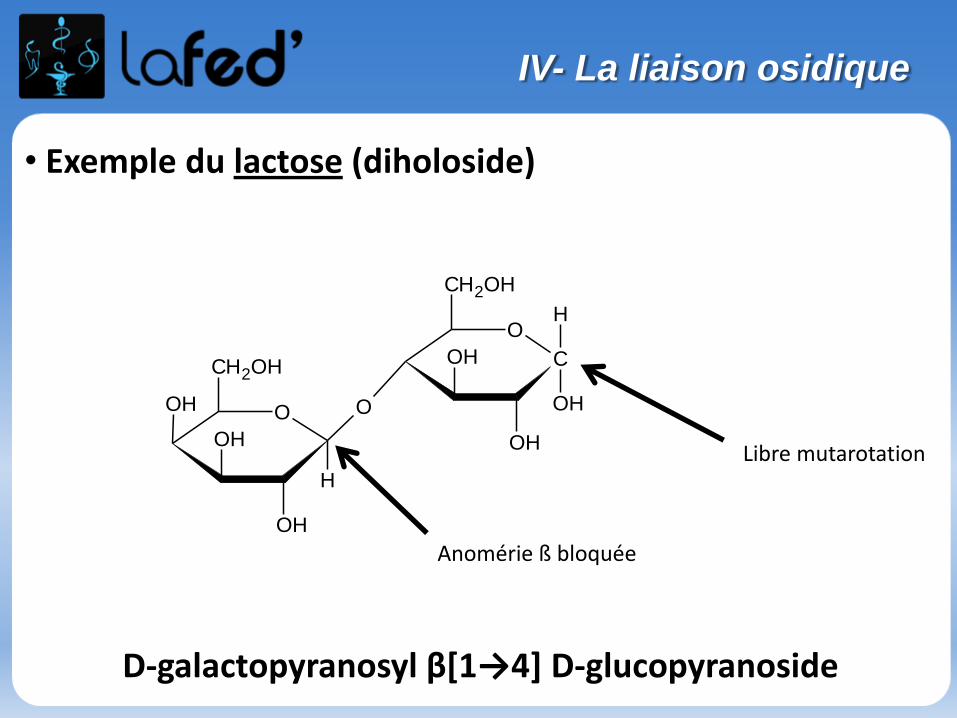
• Exemple du lactose (diholoside)
D-galactopyranosyl β*1→4+ D-glucopyranoside
Libre mutarotation
Anomérie ß bloquée
O
H
O
OH
OH
OH
CH2OH
O
C
H
OH
OH
CH2OH
OH
IV- La liaison osidique
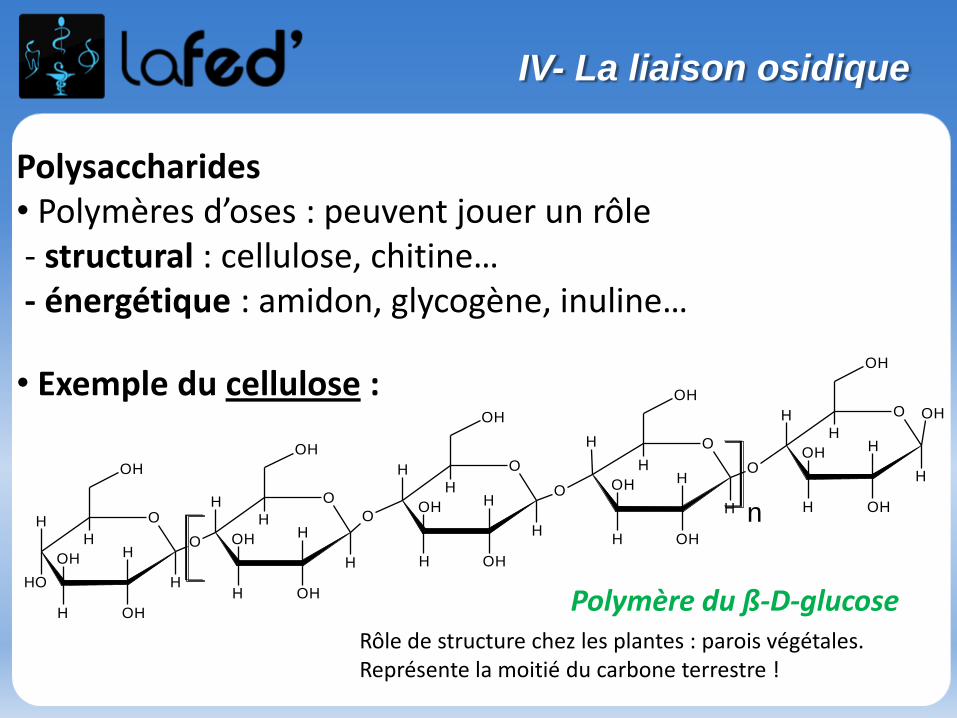
Polysaccharides• Polymères d’oses : peuvent jouer un rôle- structural : cellulose, chitine…- énergétique : amidon, glycogène, inuline…
• Exemple du cellulose :O
H
HH
H
OOH
H OH
OH
O
H
HH
H
O OH
H OH
OH
O
H
HH
H
OOH
H OH
OH
O
H
HH
H
O OH
H OH
OH
O
H
HH
H
OH
OH
H OH
OH
OH
n
Polymère du ß-D-glucoseRôle de structure chez les plantes : parois végétales.Représente la moitié du carbone terrestre !
IV- La liaison osidique

• Les osidases
Ce sont des enzymes qui catalysent l’hydrolyse des liaisons osidiques.Elles ont une spécificité relative : elles ne reconnaissent qu’unanomère en particulier.
Exemples : α-glucosidaseβ-galactosidaseβ-glucosidase (absente chez l’homme)
IV- La liaison osidique
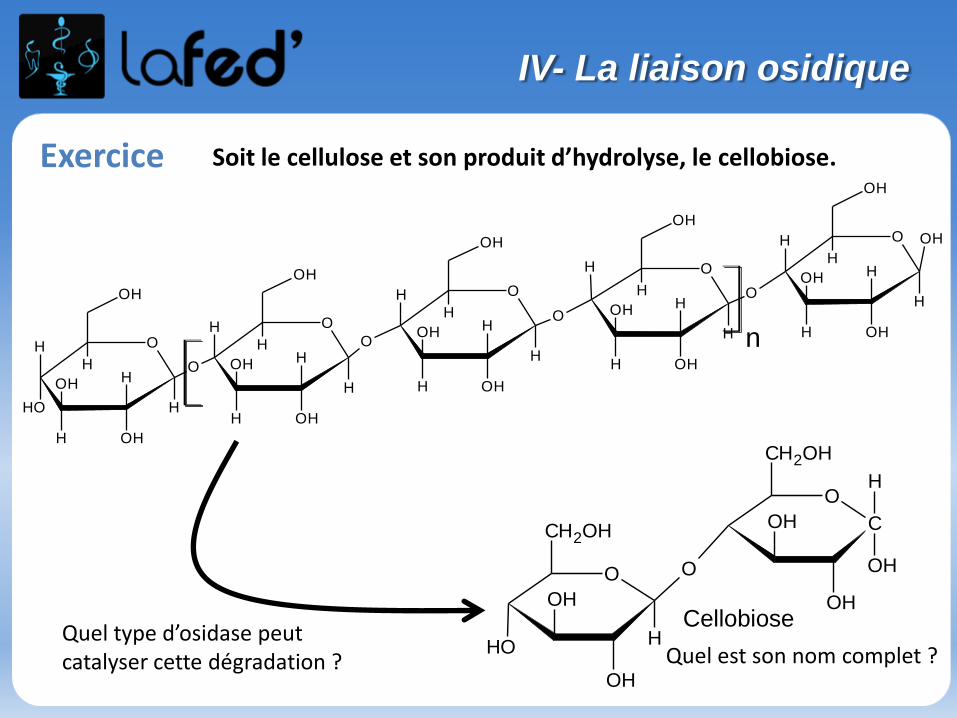
Exercice Soit le cellulose et son produit d’hydrolyse, le cellobiose.
IV- La liaison osidique
O
H
HH
H
OOH
H OH
OH
O
H
HH
H
O OH
H OH
OH
O
H
HH
H
OOH
H OH
OH
O
H
HH
H
O OH
H OH
OH
O
H
HH
H
OH
OH
H OH
OH
OH
n
CellobioseQuel type d’osidase peut catalyser cette dégradation ? Quel est son nom complet ?
O
H
O
OH
OH
OH
CH2OH
O
C
H
OH
OH
CH2OH
OH

• Par opposition aux glucides simples (holosides).
• Est appelé hétéroside une molécule contenant :- Une fraction glucidique- Une fraction non-glucidique (aglycone)
• La liaison reliant un ose à l’aglycone est généralement uneliaison acétal ou analogue à une liaison acétal.
Proportions variables
OO
ON
Aglycone Aglycone
N-hétéroside O-hétéroside
V- Glucides complexes :
hétérosides
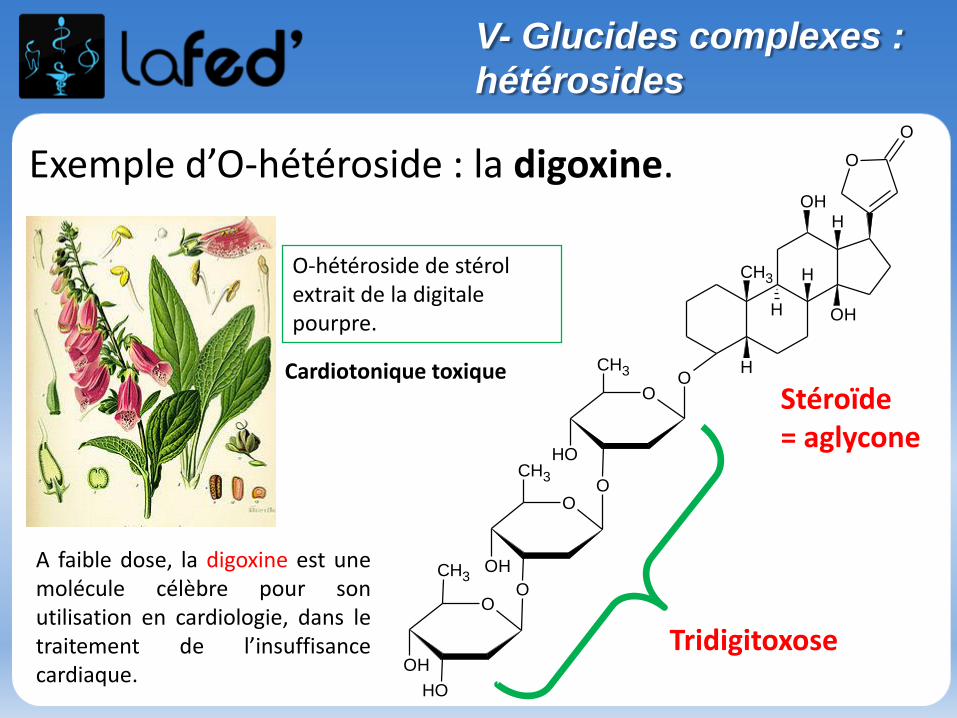
O
OH
O
CH3O
O
O
OH
H
H
H
CH3
H
O
OH
O
CH3
O
OH
OH
CH3
OH
Exemple d’O-hétéroside : la digoxine.
Tridigitoxose
Stéroïde= aglycone
O-hétéroside de stérol extrait de la digitale pourpre.
Cardiotonique toxique
A faible dose, la digoxine est unemolécule célèbre pour sonutilisation en cardiologie, dans letraitement de l’insuffisancecardiaque.
V- Glucides complexes :
hétérosides
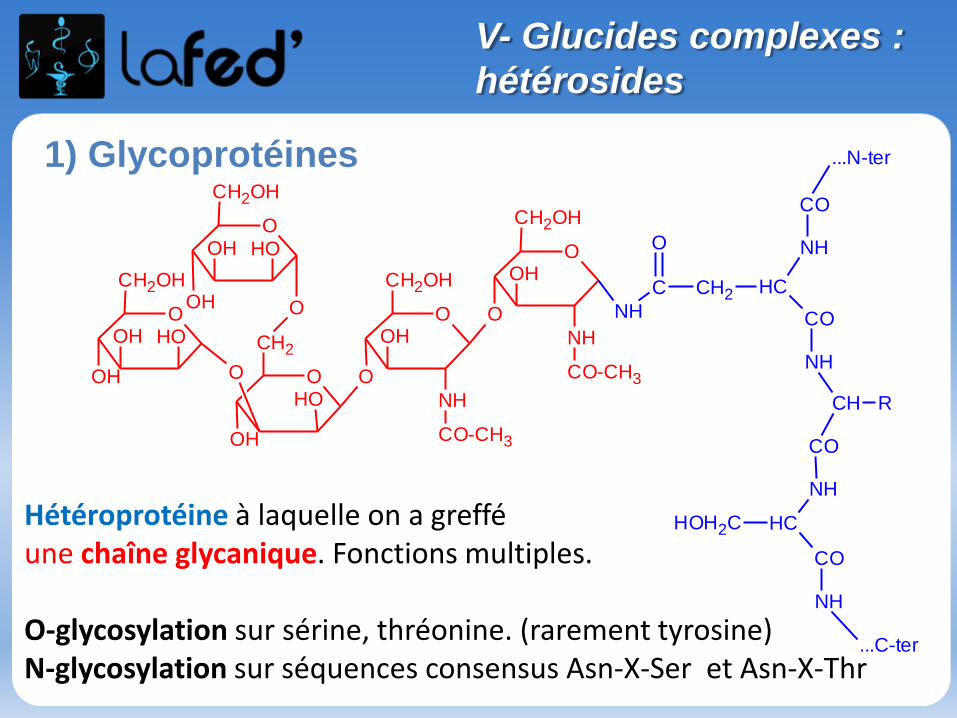
Hétéroprotéine à laquelle on a grefféune chaîne glycanique. Fonctions multiples.
O-glycosylation sur sérine, thréonine. (rarement tyrosine)N-glycosylation sur séquences consensus Asn-X-Ser et Asn-X-Thr
O
NH
OH
O
CH2OH
...N-ter
CO
NH
CH
CO
NH
CH
CO
NH
CH
CO
NH
...C-ter
CH2C
O
NH
CO-CH3
O
NH
OH
O
CH2OH
CO-CH3
OOH
O
OH
CH2
O
OOHOH
OH
CH2OH
OOHOH
OH
CH2OH
R
HOH2C
1) Glycoprotéines
V- Glucides complexes :
hétérosides

Constituants importants de la paroi de certaines bactéries.Ce sont des réseaux d’osides reliés entre eux par des petitspeptides.Ils sont la cible de certains antiobiotiques, notamment les pénicillines.
Constituants importants de la matrice extra-cellulaire.Fraction glucidique > Fraction protéique
(Contrairement aux glycoprotéines)Fonction glucidique = Glycosaminoglycanes (GAG) → « Glycanes » contenant de la glycosamine.
3) Protéoglycannes
2) Peptidoglycannes
V- Glucides complexes :
hétérosides
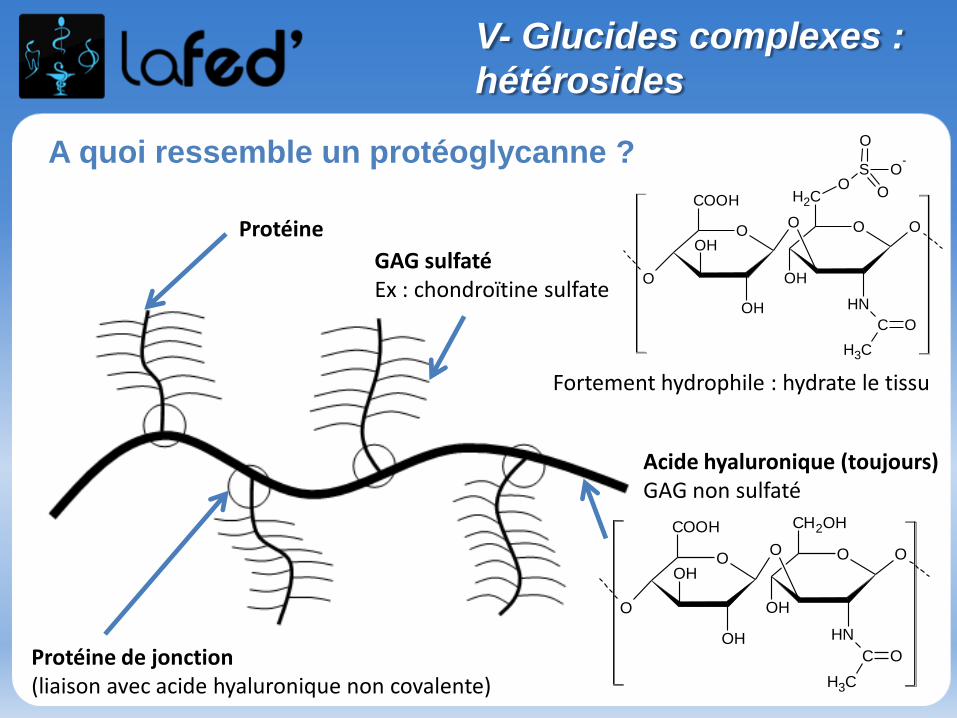
A quoi ressemble un protéoglycanne ?
Acide hyaluronique (toujours)GAG non sulfaté
Protéine
Protéine de jonction (liaison avec acide hyaluronique non covalente)
O
OH
OH
O
COOH
O
NH
CH2OH
O
OH
C
CH3
O
O
GAG sulfatéEx : chondroïtine sulfate
O
OH
OH
O
COOH
O
NH
CH2
O
OH
C
CH3
O
O
OS
O
O
O-
Fortement hydrophile : hydrate le tissu
V- Glucides complexes :
hétérosides

Stage de Pré-Rentrée 2012UE 1 – Les lipides
283
Diaporama réalisé par les tuteurs de La Fed’

Qu’est ce qu’un lipide ?
Les lipides forment l'ensemble des graisses. Caractère onctueux, pateux.
Substances biologiques solubles dans les solvants organiques (CH3OH, chloroforme…) = lipophilie
Très peu solubles dans l’eau = hydrophobie
Cires(solides)
Huiles (liquides)
Augmentation en température
Pates
I- Généralités
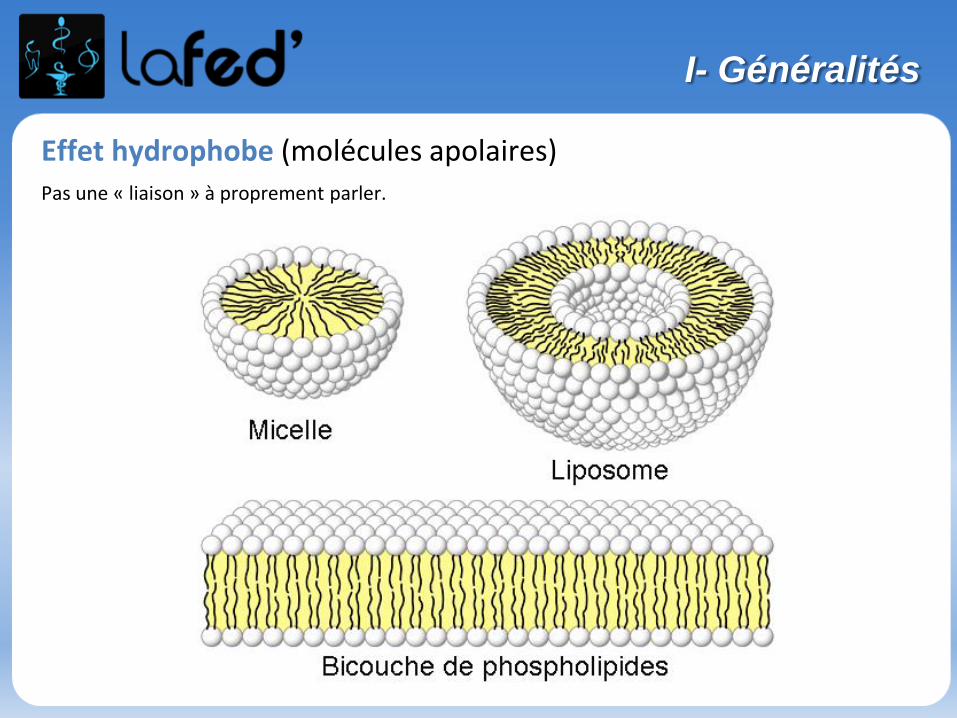
Effet hydrophobe (molécules apolaires)Pas une « liaison » à proprement parler.
I- Généralités
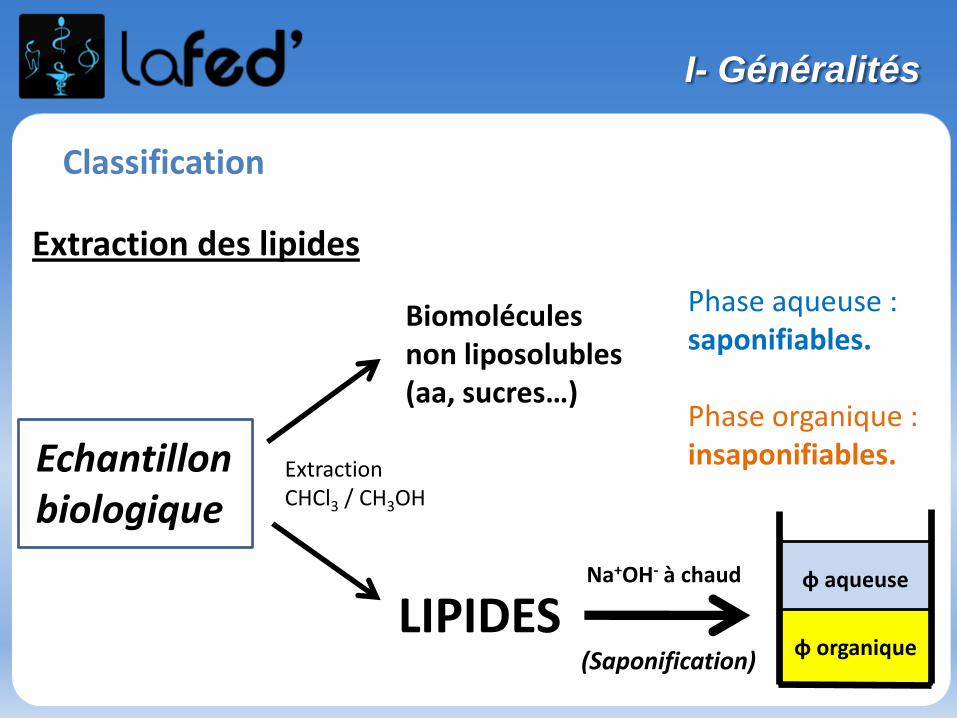
φ aqueuse
φ organique
Classification
Extraction des lipides
Echantillon biologique
ExtractionCHCl3 / CH3OH
Biomolécules non liposolubles(aa, sucres…)
LIPIDESNa+OH- à chaud
(Saponification)
Phase aqueuse : saponifiables.
Phase organique : insaponifiables.
I- Généralités

Fonction des lipides
Comme pour les protides et les glucides, leur fonction est multiple !
Forme des feuillets lipidiques et membranes
Ex : Organisation en bicouche lipidique des membranes grâce à leur caractère amphipatique
Communications :
Rôle de transduction du signal (Milieu EC vers Milieu IC)
Hormones stéroïdes
Etc…
Réserve énergétique
I- Généralités

Exercices
Connaissez-vous des lipides « stéroïdes » :- Qui sont des hormones sexuelles ?- Qui doivent être contrôlés dans l’apport alimentaire ?
QCM : Les items suivant sont-ils « Vraaaaiiiis » ou « Faaaauuuux » A. Les lipides se caractérisent par leur caractère hydrophile.B. Le cœur d’une micelle est aqueux.C. Les lipides sont souvent classés en fonction de leur réponse à la réaction
d’estérification. D. Les lipides sont une réserve d’énergie importante pour l’organisme. E. Les lipides peuvent jouer un rôle dans la communication entre cellules.
I- Généralités
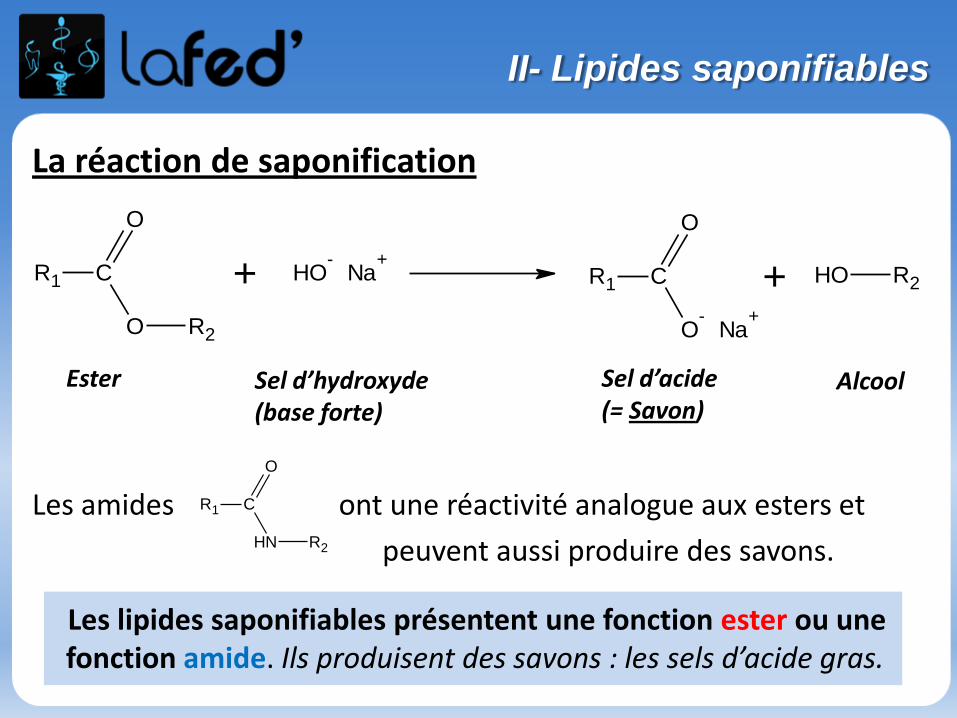
La réaction de saponification
Les amides ont une réactivité analogue aux esters et
peuvent aussi produire des savons.
Les lipides saponifiables présentent une fonction ester ou une fonction amide. Ils produisent des savons : les sels d’acide gras.
R1 C
O
O R2
+ OH-
Na+
R1 C
O
O-
Na+
+ OH R2
Ester Sel d’hydroxyde(base forte)
Sel d’acide(= Savon)
Alcool
R1 C
O
NH R2
II- Lipides saponifiables

→ d’acides grasCe sont des acides monocarboxyliques R-COOHR est une chaîne aliphatique non ramifiée comportant au moins 4 C.
Elle peut être saturée ou insaturée (mono ou poly).
→ de glycérol ou d’un alcool gras ou d’un stérolEstérifient les acides gras. Si glycérol : glycéride si alcool gras : céride si stérol : stéride
Les Lipides simples
• Ils sont formés à partir :
II- Lipides saponifiables

1) Les acides gras
Nomenclature usuelle des AG :
Noms triviaux, différents de ceux utilisés en chimie organique.
Quelques-uns sont à connaitre :
CH3–CH2–CH2–COOH Acide butyrique (4 carbones)
CH3–(CH2)12–COOH Acide myristique (14 carbones)
CH3–(CH2)14–COOH Acide palmitique (16 carbones)
CH3–(CH2)16–COOH Acide stéarique (18 carbones) etc…
II- Lipides saponifiables

Nomenclature physiologique des AG :
Cn:x(n-a) ou Cn:x(ωa)
n = nombre de carbone
x = nombre de double liaison (= d.l.)
a = atome de carbone portant la première d.l. à partir du CH3 terminal.
Intérêt : permet de classer les AG en « familles d’AG »
rôles fonctionnels différents (ω9, ω6, ω3…)
II- Lipides saponifiables
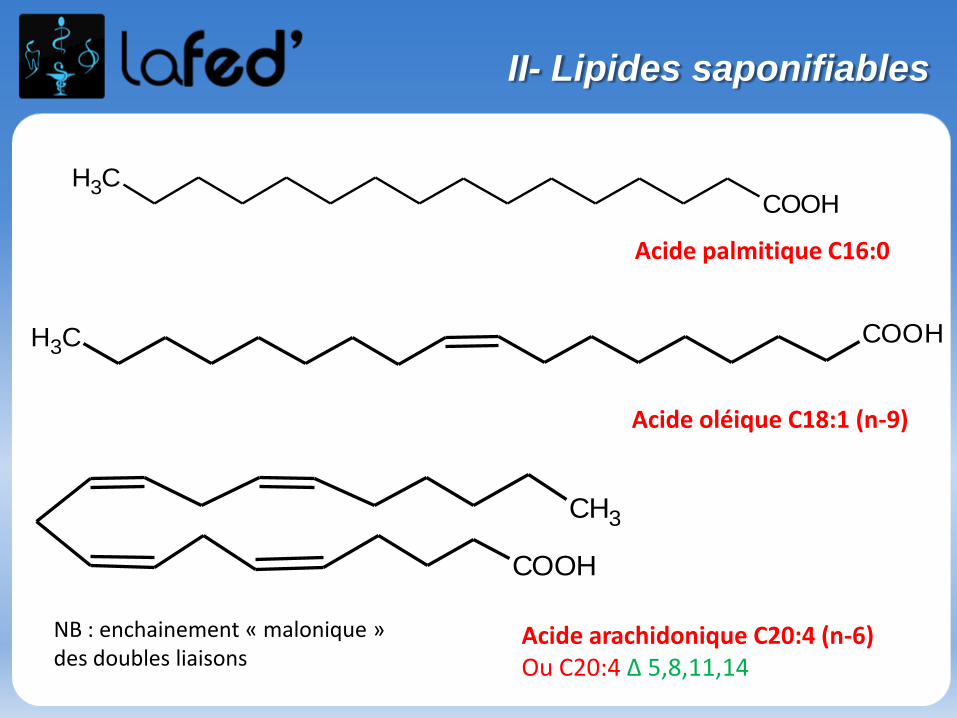
Acide palmitique C16:0
CH3COOH
CH3COOH
Acide oléique C18:1 (n-9)
CH3
COOH
Acide arachidonique C20:4 (n-6)Ou C20:4 Δ 5,8,11,14
NB : enchainement « malonique » des doubles liaisons
II- Lipides saponifiables

Température de fusion Tf des acides gras
Tf44,2°C 52,0°C 69,6°C 75,4°C63,1°C
C12:0 C14:0 C16:0 C18:0 C20:0
Augmente avec le poids moléculaire (nombre de carbone)
Diminue avec le nombre d’insaturations (= la fluidité augmente !)
C18:0C18:1
Tf13,4°C 69,6°C
C18:2
« Plus c’est long et dur… plus on doit chauffer ! »
-9°C-17°C
C18:3
II- Lipides saponifiables
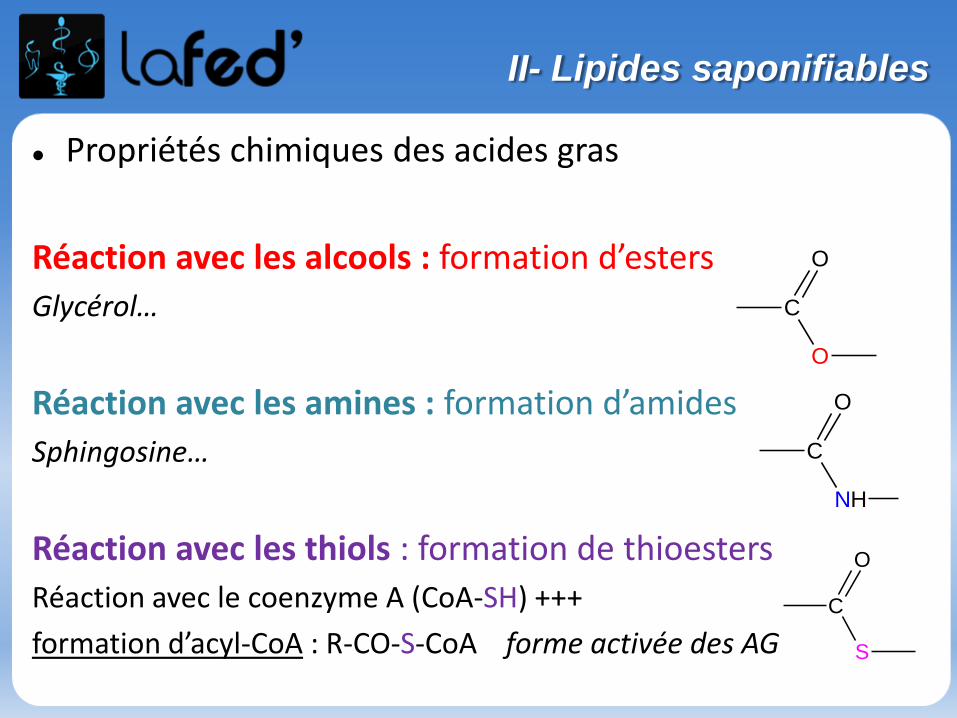
Propriétés chimiques des acides gras
Réaction avec les alcools : formation d’esters
Glycérol…
Réaction avec les amines : formation d’amides
Sphingosine…
Réaction avec les thiols : formation de thioesters
Réaction avec le coenzyme A (CoA-SH) +++
formation d’acyl-CoA : R-CO-S-CoA forme activée des AG
C
O
O
C
O
NH
C
O
S
II- Lipides saponifiables

Notions de métabolisme des AG
Dégradation : β-oxydation
Voie métabolique mitochondriale
Récupère des acides gras : Acétyl CoA
Pouvoir réducteur (NADH, FADH2)
Synthèse (jusqu’à C16:0) : Enzyme « AG synthase »
Voie métabolique cytosolique
Consommation de pouvoir réducteur (NADPH, FADH2)
Elongation à partir d’Acétyl-CoA (C2) → AG nombre pair de C.
II- Lipides saponifiables
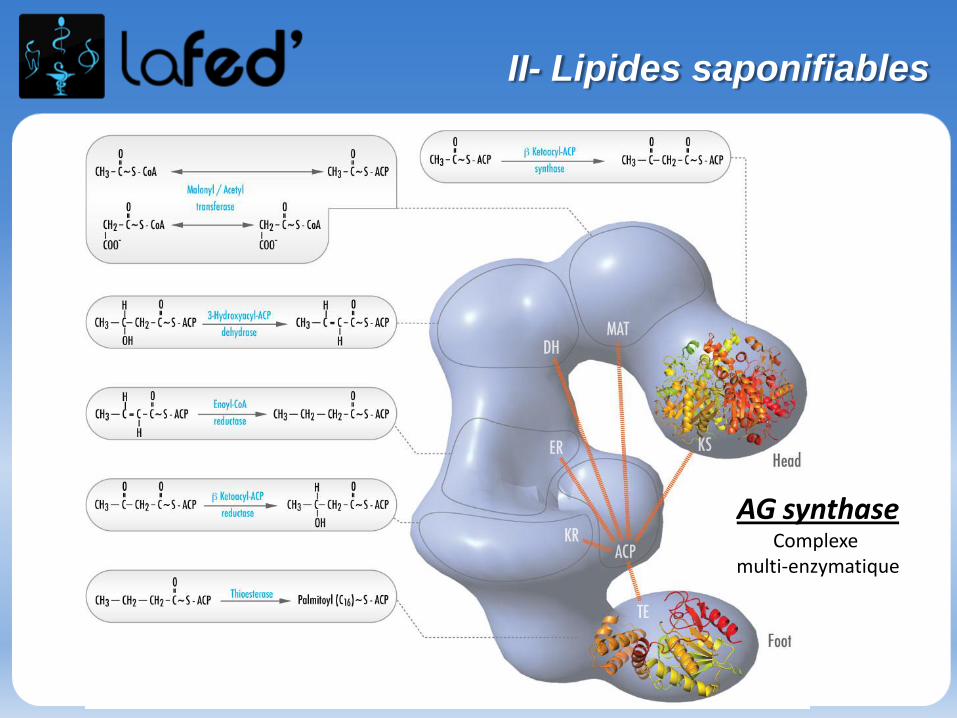
AG synthaseComplexe
multi-enzymatique
II- Lipides saponifiables
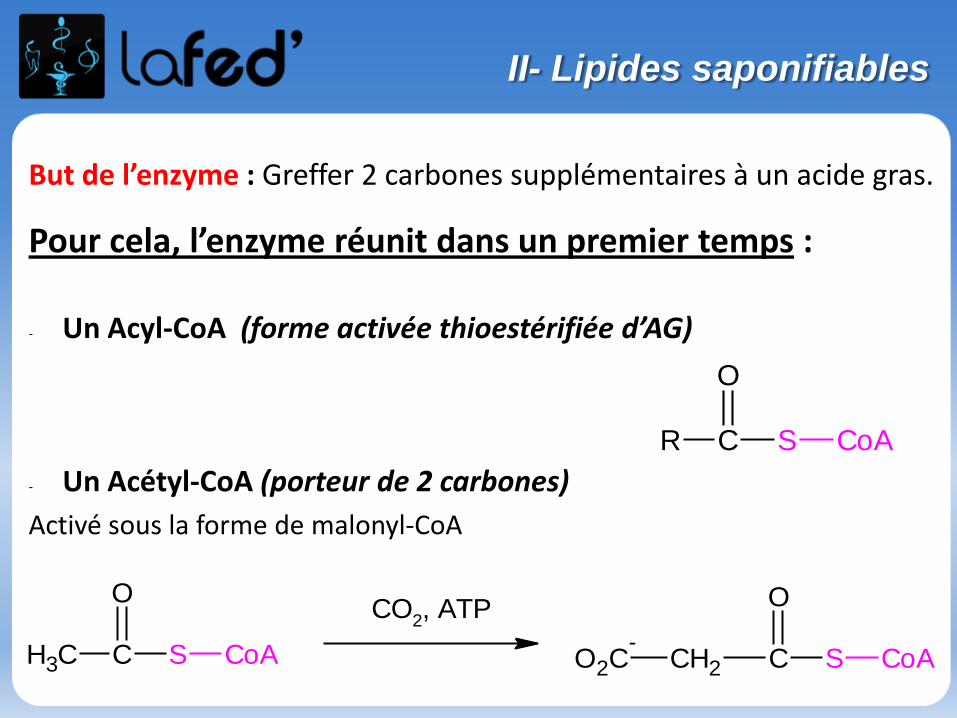
But de l’enzyme : Greffer 2 carbones supplémentaires à un acide gras.
Pour cela, l’enzyme réunit dans un premier temps :
- Un Acyl-CoA (forme activée thioestérifiée d’AG)
- Un Acétyl-CoA (porteur de 2 carbones)
Activé sous la forme de malonyl-CoA
R C S
O
CoA
CH3 C S
O
CoA CH2 C S
O
CoAO2C-
CO2, ATP
II- Lipides saponifiables
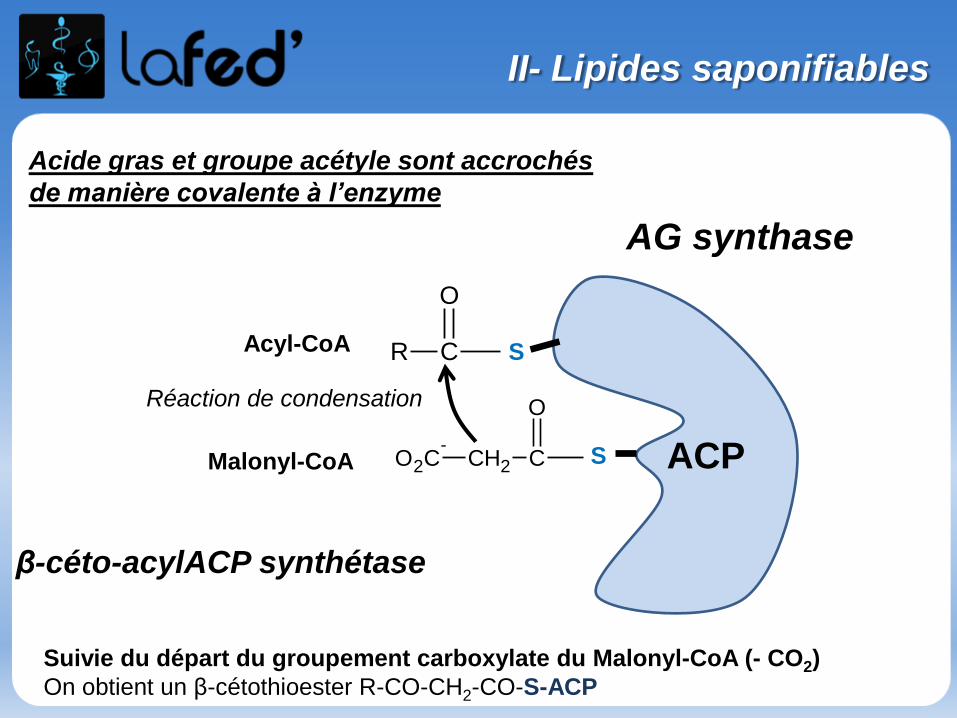
ACPS
S
AG synthase
Acide gras et groupe acétyle sont accrochés
de manière covalente à l’enzyme
R C
O
O2C-
CH2 C
O
Acyl-CoA
Malonyl-CoA
Réaction de condensation
Suivie du départ du groupement carboxylate du Malonyl-CoA (- CO2)
On obtient un β-cétothioester R-CO-CH2-CO-S-ACP
β-céto-acylACP synthétase
II- Lipides saponifiables
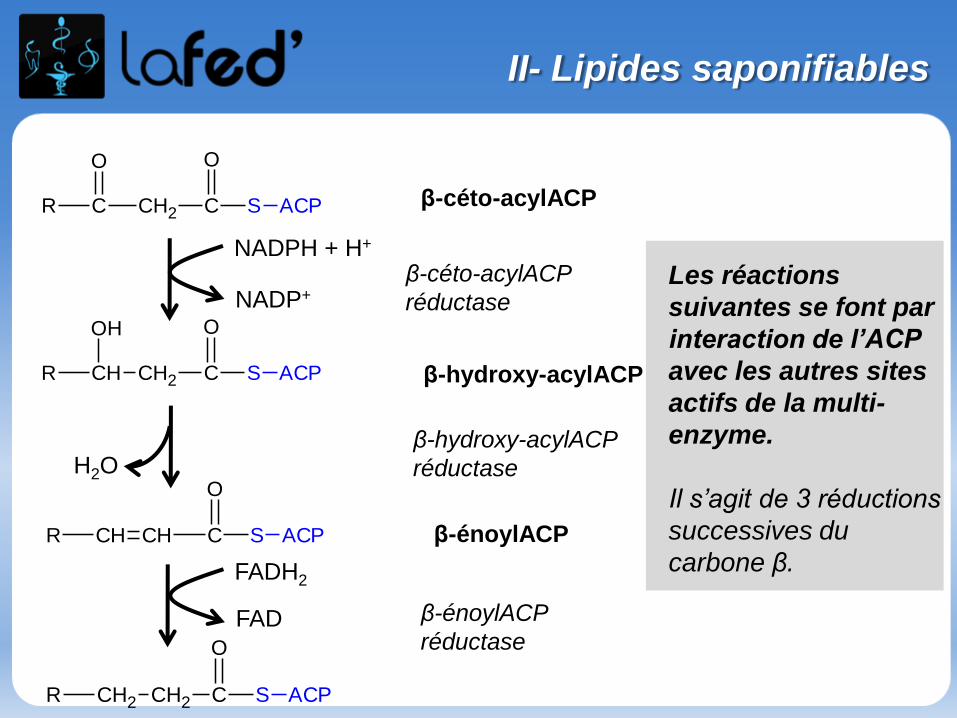
Les réactions
suivantes se font par
interaction de l’ACP
avec les autres sites
actifs de la multi-
enzyme.
Il s’agit de 3 réductions
successives du
carbone β.
CH2 C S
O
ACPC
O
R
CH2 C S
O
ACPCH
OH
R
CH C S
O
ACPCHR
CH2 C S
O
ACPCH2R
NADPH + H+
NADP+
FADH2
FAD
H2O
β-céto-acylACP
β-hydroxy-acylACP
β-énoylACP
β-céto-acylACP
réductase
β-hydroxy-acylACP
réductase
β-énoylACP
réductase
II- Lipides saponifiables
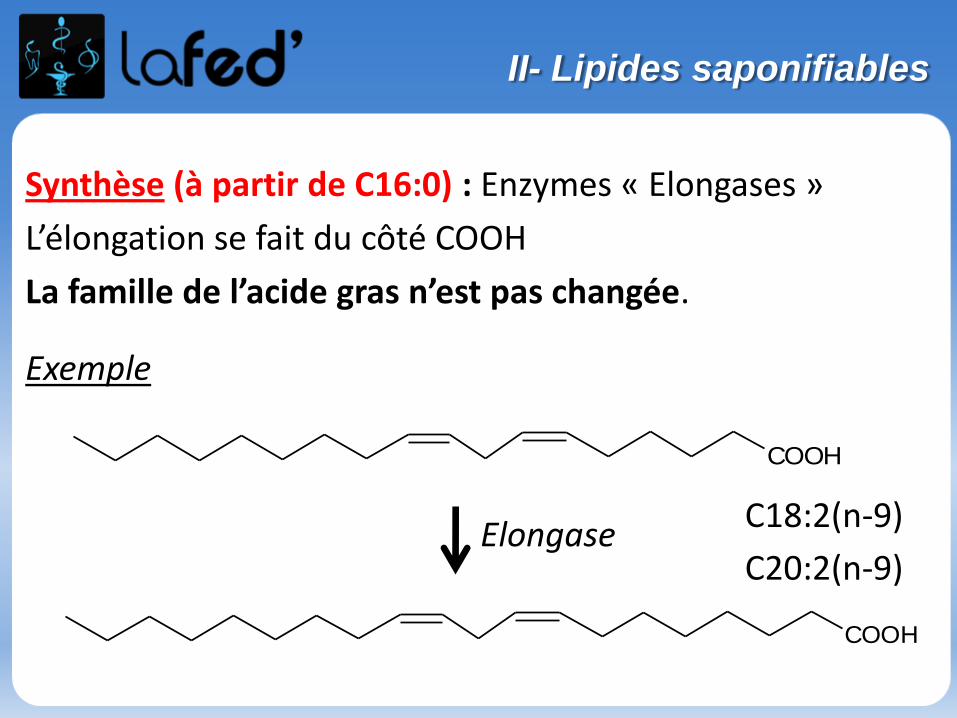
Synthèse (à partir de C16:0) : Enzymes « Elongases »
L’élongation se fait du côté COOH
La famille de l’acide gras n’est pas changée.
Exemple
ElongaseC18:2(n-9)
C20:2(n-9)
II- Lipides saponifiables
COOH
COOH
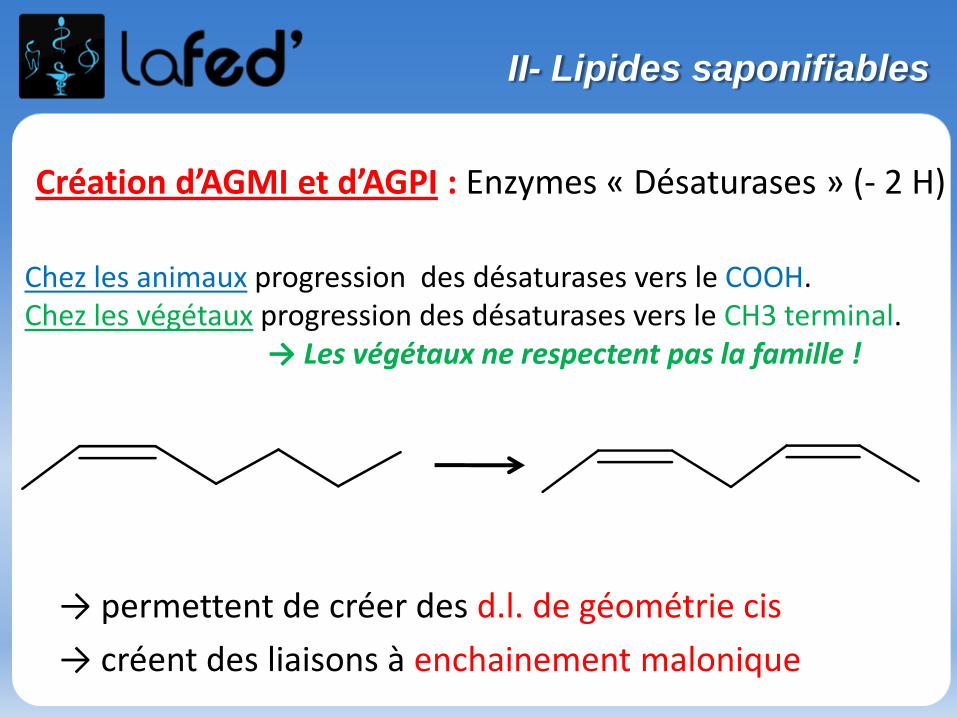
Création d’AGMI et d’AGPI : Enzymes « Désaturases » (- 2 H)
Chez les animaux progression des désaturases vers le COOH. Chez les végétaux progression des désaturases vers le CH3 terminal.
→ Les végétaux ne respectent pas la famille !
→ permettent de créer des d.l. de géométrie cis
→ créent des liaisons à enchainement malonique
II- Lipides saponifiables
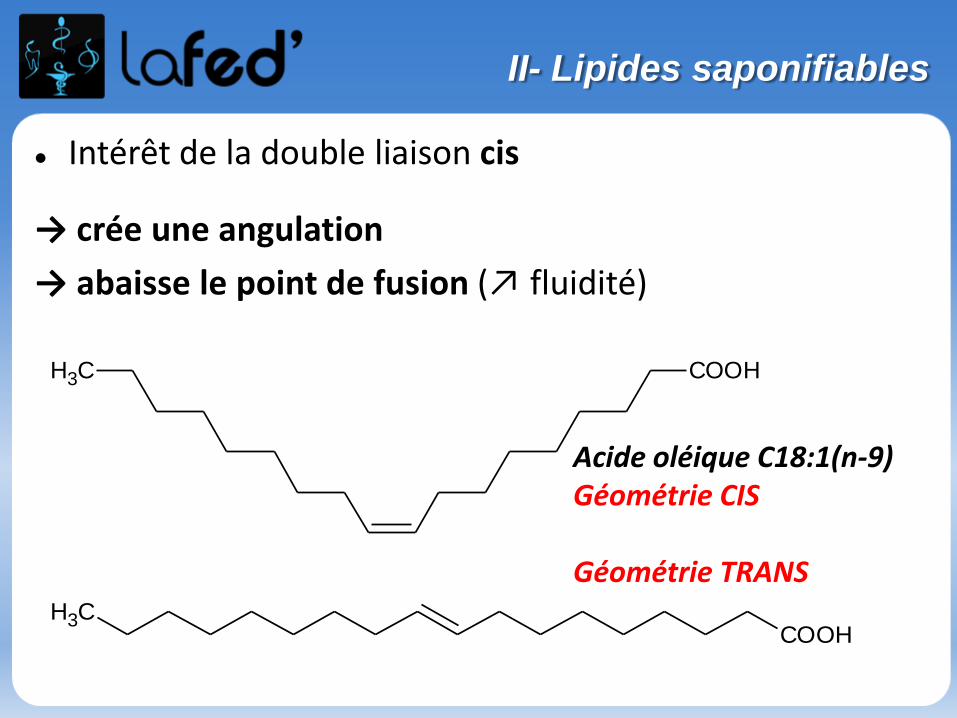
Intérêt de la double liaison cis
→ crée une angulation
→ abaisse le point de fusion (↗ fluidité)
CH3 COOH
CH3COOH
Acide oléique C18:1(n-9)Géométrie CIS
Géométrie TRANS
II- Lipides saponifiables
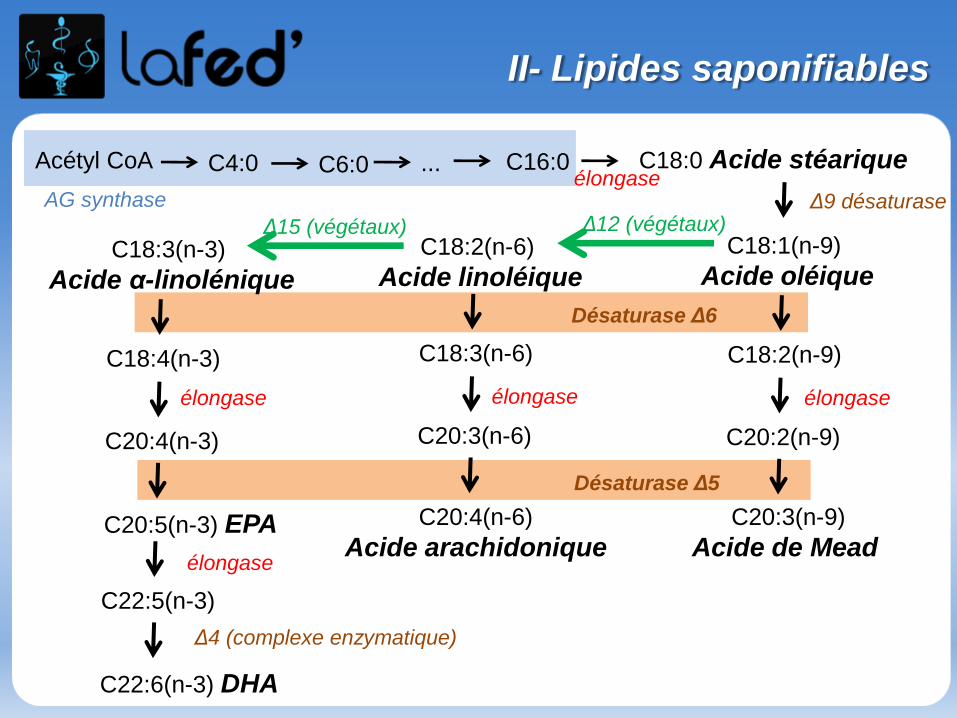
Acétyl CoA C4:0 C6:0 ... C16:0 C18:0 Acide stéarique
C18:1(n-9)
Acide oléique
élongase
C18:2(n-6)
Acide linoléiqueC18:3(n-3)
Acide α-linolénique
C18:2(n-9)
C20:2(n-9)
C20:3(n-9)
Acide de Mead
C18:3(n-6)
C20:3(n-6)
C20:4(n-6)
Acide arachidonique
C18:4(n-3)
C20:4(n-3)
C20:5(n-3) EPA
élongaseélongaseélongase
C22:5(n-3)
C22:6(n-3) DHA
AG synthase
élongase
Désaturase Δ6
Désaturase Δ5
Δ4 (complexe enzymatique)
Δ9 désaturaseΔ12 (végétaux)Δ15 (végétaux)
II- Lipides saponifiables

Glycérol
Les acides gras ne sont quasiment jamais retrouvés sous forme libre.
Graisses et huiles que l’on retrouve dans les plantes et les animaux :
triglycérides (= triacylglycérol)
Ce sont des « triesters » d’acide gras OH
OH
OH CH2
CH
CH2
b) Association avec le glycérol
II- Lipides saponifiables
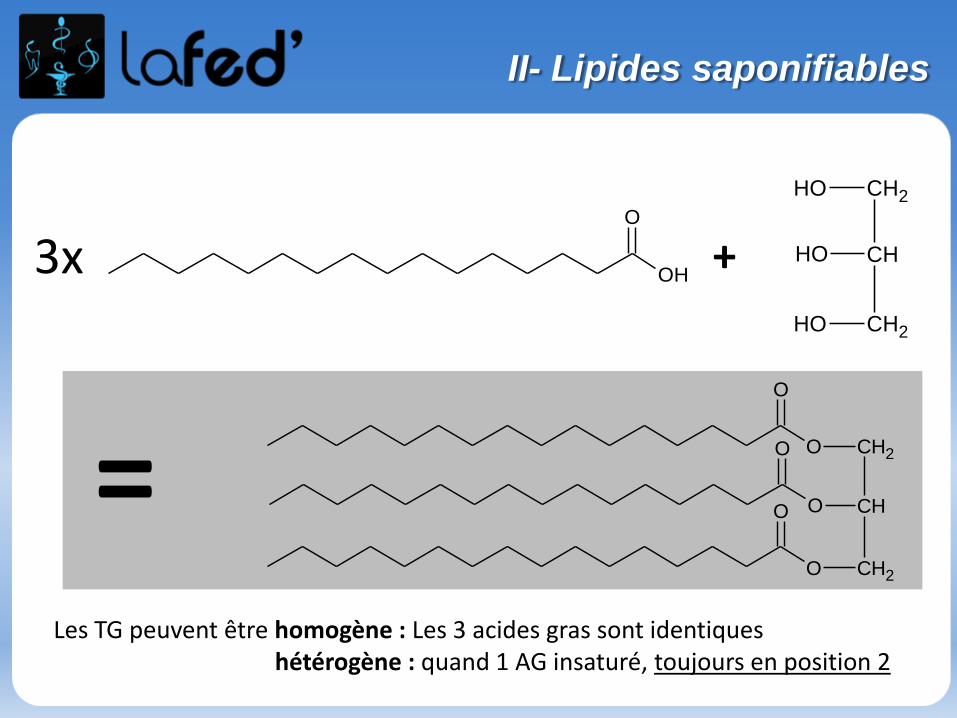
OH
OH
OH CH2
CH
CH2
O
OH3x +
O
O
O CH2
CH
CH2
O
O
O=Les TG peuvent être homogène : Les 3 acides gras sont identiques
hétérogène : quand 1 AG insaturé, toujours en position 2
II- Lipides saponifiables

Graisses et huiles alimentaires : mélanges complexes de triglycérides simples et mixtes.
Beurre : solide à température ambianteMajoritairement triglycérides saturés (Tf haut)
Huiles : liquide à température ambiante+ riches en triglycérides insaturés (Tf bas)
Huile d’olive : TG homogènes à acide oléique +++
II- Lipides saponifiables
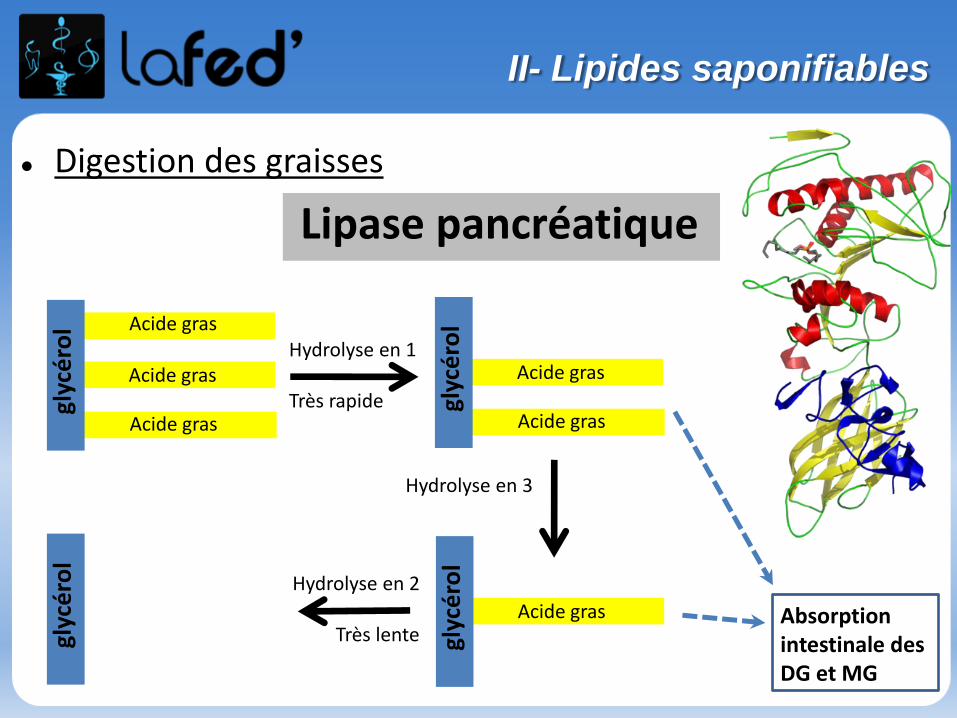
Digestion des graisses
Lipase pancréatique
Acide gras
Acide gras
Acide gras
glyc
éro
l
Acide gras
Acide gras
glyc
éro
l
Acide gras
glyc
éro
l
glyc
éro
l
Hydrolyse en 1
Très rapide
Hydrolyse en 2
Très lente
Hydrolyse en 3
Absorption intestinale des DG et MG
II- Lipides saponifiables

Rôle physiologique des triglycérides
Réserve énergétique +++
Stockage:
Adipocytes du tissu adipeux →
Hépatocyte
Sous forme de vacuoles lipidiques
Forme de transport plasmatique
Après absorption des MG et DG au niveau de l’intestin.
→ Ré-estérification en TG par les cellules de l’intestin.
Transport dans le plasma des TG par les lipoprotéines.
II- Lipides saponifiables

Exercices Concernant le lipide suivant
Comment appelle-t-on ce type de lipide ? Quel est son nom en nomenclature physiologique ?Les insaturations diminuent-elles ou augmentent-elles la fluidité ?Est-il synthétisé par l’homme ?
COOHCH3
Comment appelle-t-on la voie métabolique de dégradation des acides gras ?(Vous vous souvenez dans quel organite elle a lieu ?)
A partir de quel nombre de carbones l’AG synthase ne prend-t-elle plus en charge les acides gras ?
Pourquoi les acides gras ont-ils toujours un nombre pair de carbone ?
II- Lipides saponifiables
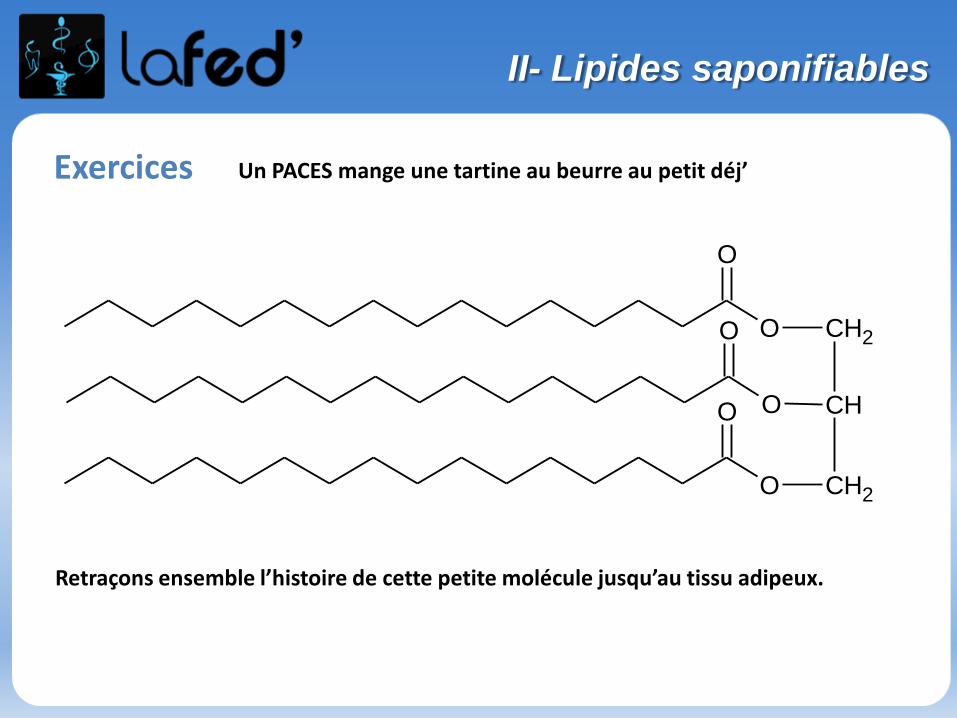
Exercices Un PACES mange une tartine au beurre au petit déj’
O
O
O CH2
CH
CH2
O
O
O
Retraçons ensemble l’histoire de cette petite molécule jusqu’au tissu adipeux.
II- Lipides saponifiables

Lipides simples : Contiennent éléments C, O, H
Lipides complexes : Contiennent d’autres éléments, tels
que de l’azote ou du phosphore.
Les lipides complexes sont impliqués dans
l’édification des membranes biologiques.
Les lipides complexes
II- Lipides saponifiables

Ils ont une structure commune:
- 2 chaînes hydrophobes
- Plate forme d'ancrage des AG et des têtes polaires
Glycérol ou sphingosine
- Tête polaire: Phosphate + Alcool
ou Ose/chaîne polyosidique
Molécules amphipatiques
Plate forme d’ancrage
2 longues chaînes hydrophobes
Tête polaire
II- Lipides saponifiables
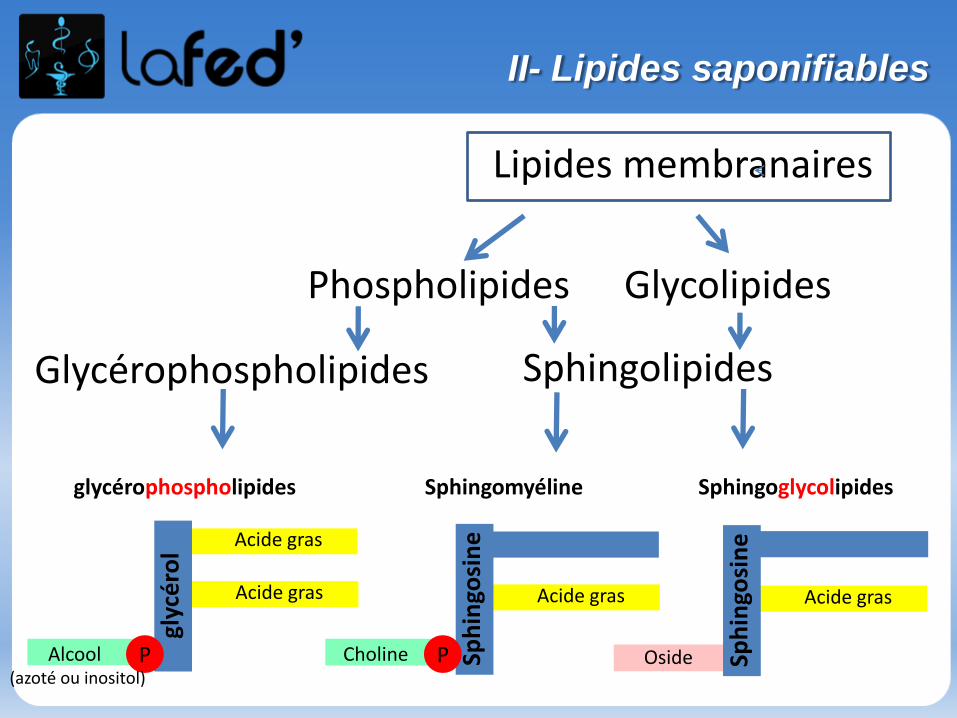
glyc
éro
l Acide gras
Acide gras
PAlcool(azoté ou inositol)
Sph
ingo
sin
e
Acide gras
OsideSph
ingo
sin
e
Acide gras
PCholine
glycérophospholipides Sphingomyéline Sphingoglycolipides
Lipides membranaires
Phospholipides Glycolipides
Glycérophospholipides Sphingolipides
II- Lipides saponifiables

On ne développera que les glycérophospholipides :
glyc
éro
l Acide gras
Acide gras
PAlcool(azoté ou inositol)
II- Lipides saponifiables
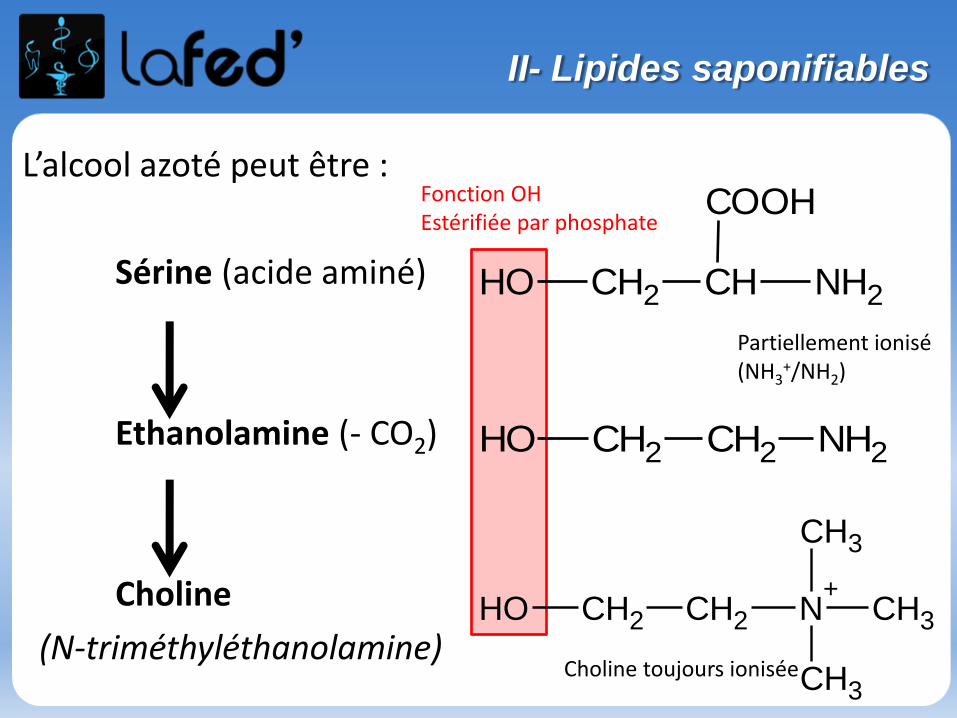
L’alcool azoté peut être :
Sérine (acide aminé)
Ethanolamine (- CO2)
Choline
(N-triméthyléthanolamine)
OH CH2 CH NH2
COOH
OH CH2 CH2 NH2
OH CH2 CH2 N+
CH3
CH3
CH3
Fonction OH Estérifiée par phosphate
Partiellement ionisé(NH3
+/NH2)
Choline toujours ionisée
II- Lipides saponifiables
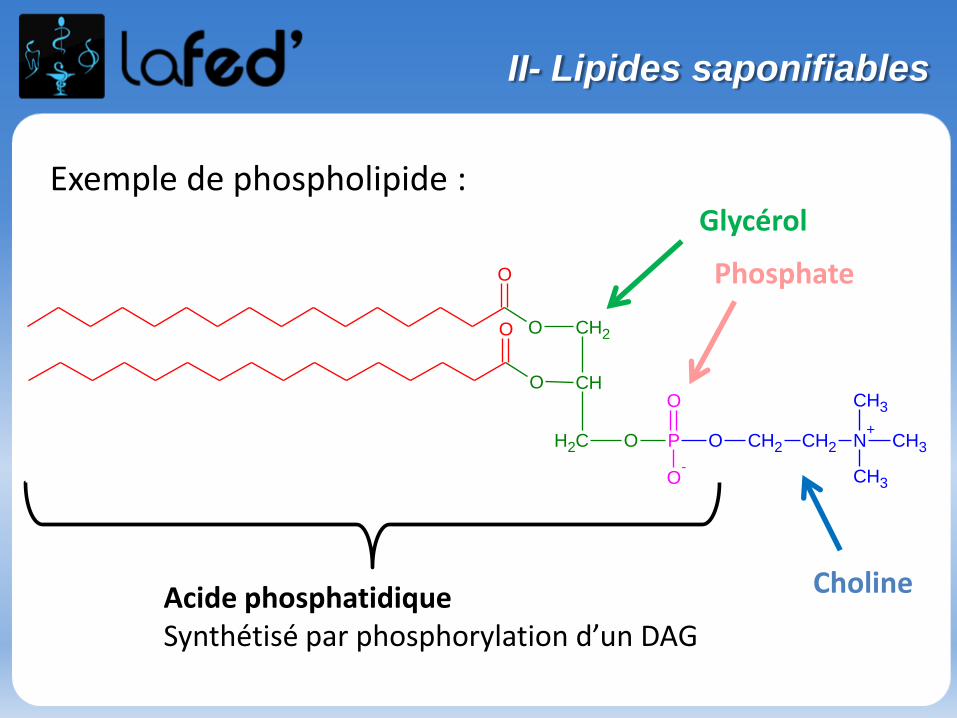
Exemple de phospholipide :
O
O
O CH2
CH
CH2
O
O
P
O
O-
O CH2 CH2 N+
CH3
CH3
CH3
Glycérol
Phosphate
CholineAcide phosphatidiqueSynthétisé par phosphorylation d’un DAG
II- Lipides saponifiables

C’est quoi la différence entre un lipide « simple » et un lipide « complexe » ?(Définition)
Exercices
Soit le schéma de glycérophospholipide suivant
Associer chacun des composantssuivants à la couleur correspondantedans le schéma :
- Glycérol- Phosphate- Choline- Acide gras saturé- Acide gras insaturé (s’il existe, quelleposition préférentielle : 1 ou 2 ?)
Position 1
Position 2
Comment appelle-t-on cette partie là ?
II- Lipides saponifiables
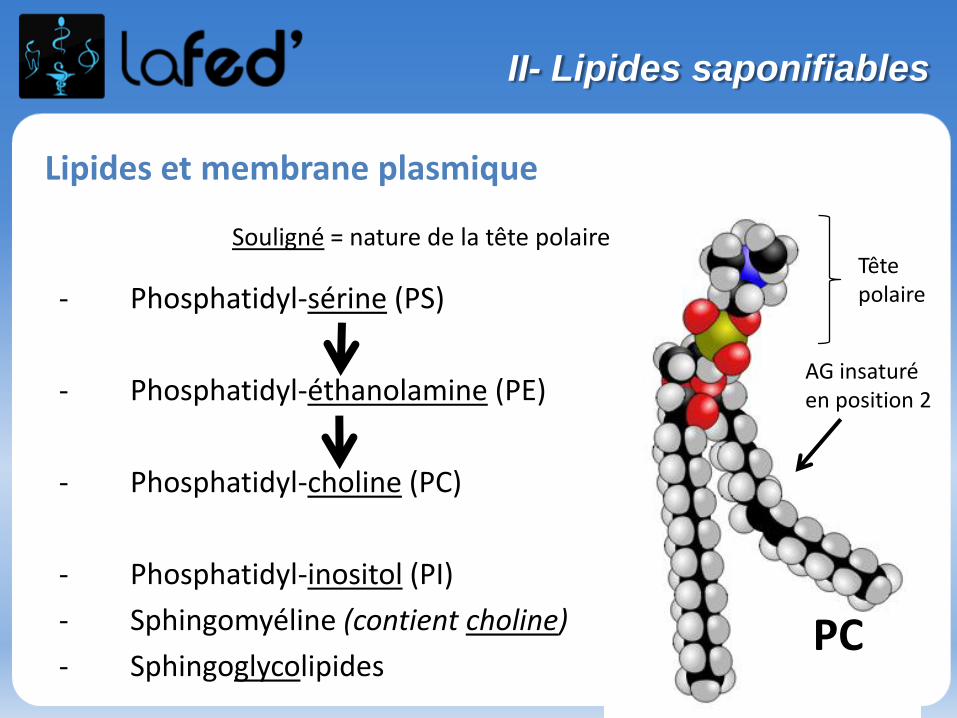
- Phosphatidyl-sérine (PS)
- Phosphatidyl-éthanolamine (PE)
- Phosphatidyl-choline (PC)
- Phosphatidyl-inositol (PI)
- Sphingomyéline (contient choline)
- Sphingoglycolipides
Souligné = nature de la tête polaire
PC
AG insaturé en position 2
Tête polaire
Lipides et membrane plasmique
II- Lipides saponifiables

Les lipides complexes sont des molécules amphipatiques.→ présentent un pôle hydrophile et un pôle hydrophobe.
En milieu aqueux, on observe une tendance spontanée à laformation d’agrégats lipidiques, pour protéger le pôlehydrophobe du solvant.
II- Lipides saponifiables
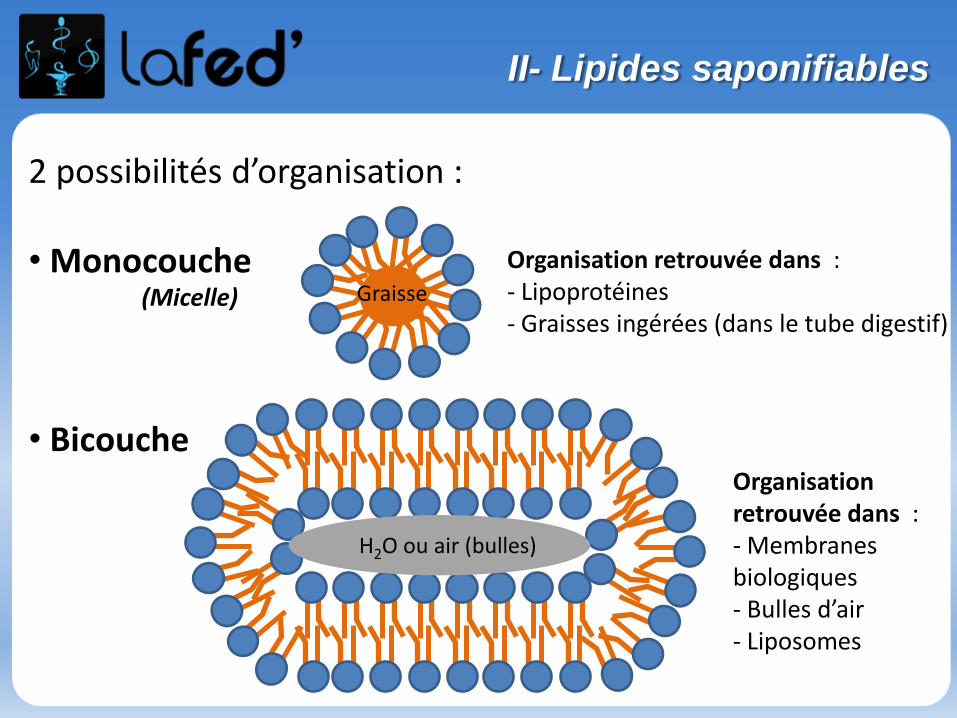
2 possibilités d’organisation :
• Monocouche
• Bicouche
Graisse(Micelle)
Organisation retrouvée dans :- Lipoprotéines- Graisses ingérées (dans le tube digestif)
H2O ou air (bulles)
Organisation retrouvée dans :- Membranes biologiques- Bulles d’air- Liposomes
II- Lipides saponifiables
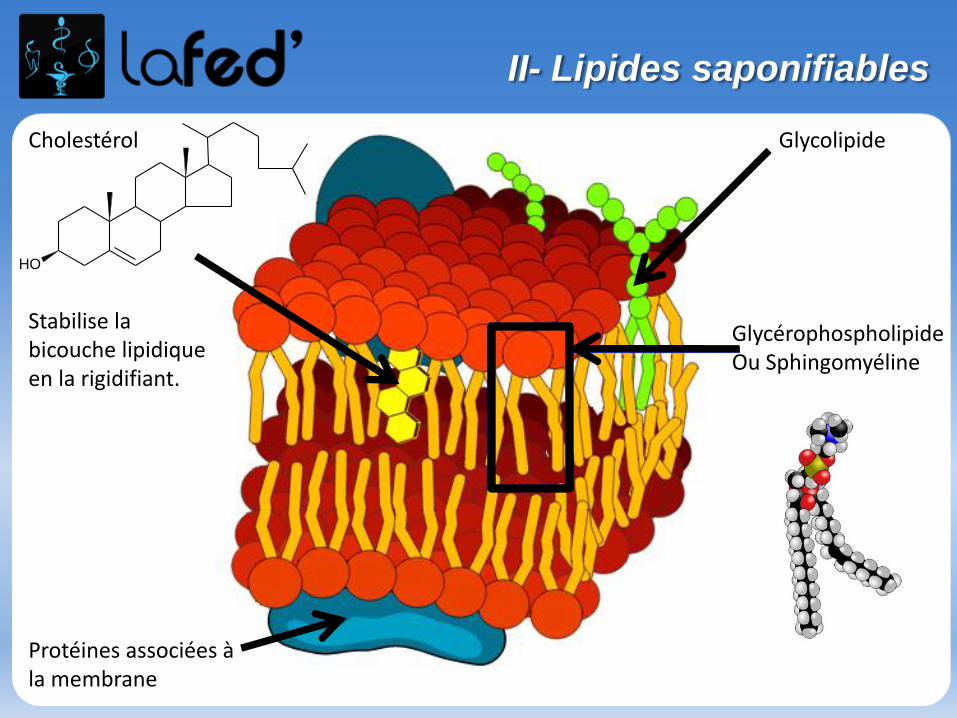
OH
Cholestérol Glycolipide
Protéines associées à la membrane
GlycérophospholipideOu Sphingomyéline
Stabilise la bicouche lipidique en la rigidifiant.
II- Lipides saponifiables
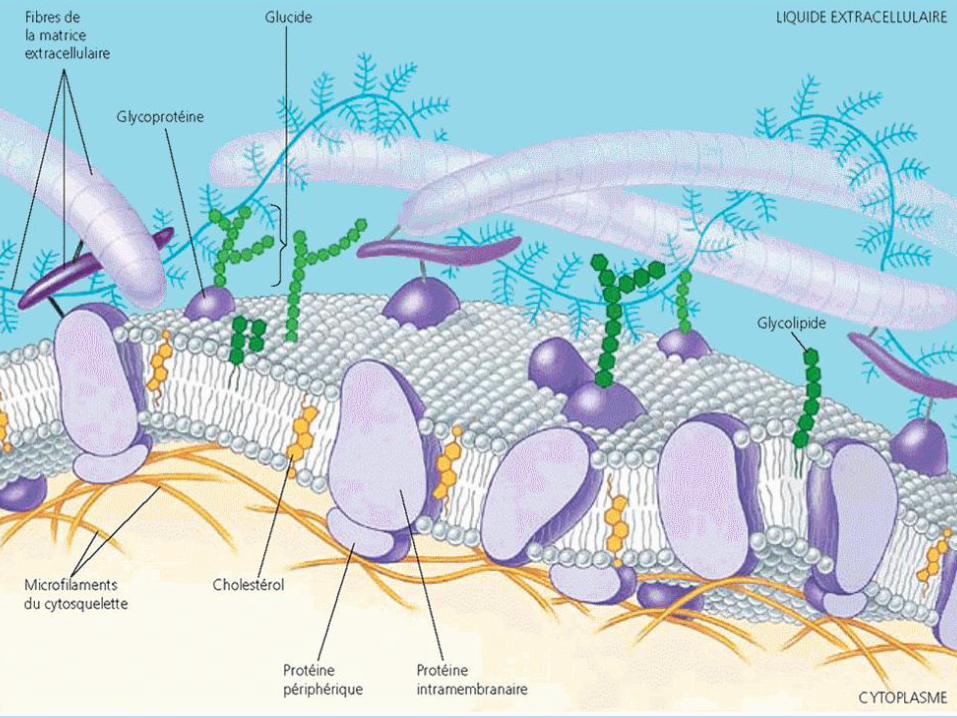
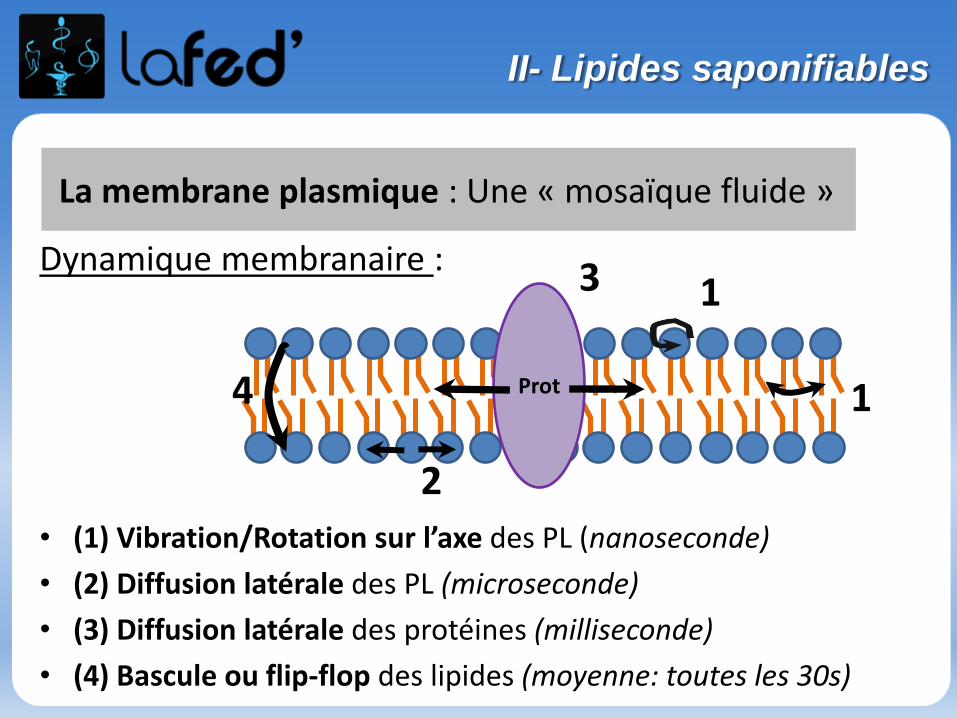
Dynamique membranaire :
• (1) Vibration/Rotation sur l’axe des PL (nanoseconde)
• (2) Diffusion latérale des PL (microseconde)
• (3) Diffusion latérale des protéines (milliseconde)
• (4) Bascule ou flip-flop des lipides (moyenne: toutes les 30s)
La membrane plasmique : Une « mosaïque fluide »
1
4 Prot 1
2
3
II- Lipides saponifiables
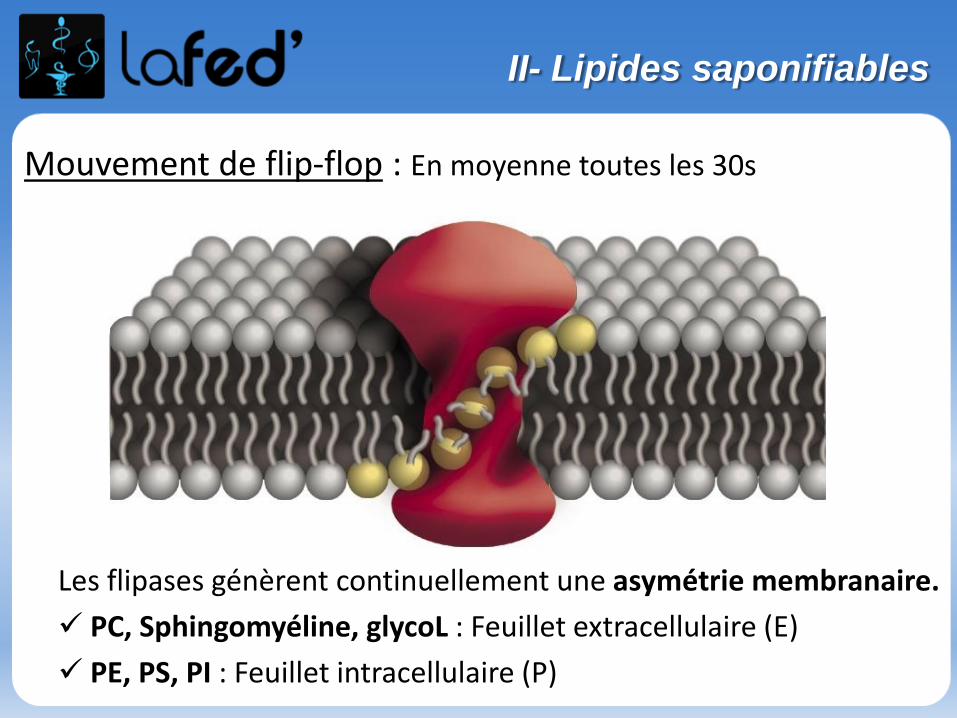
Mouvement de flip-flop : En moyenne toutes les 30s
Les flipases génèrent continuellement une asymétrie membranaire.
PC, Sphingomyéline, glycoL : Feuillet extracellulaire (E)
PE, PS, PI : Feuillet intracellulaire (P)
II- Lipides saponifiables
Exercice
QCM : Répondre vrai ou faux, comme d’habitude.
A. L’insaturation « cis » des glycérophospholipides permet de rigidifier la membrane.B. La membrane plasmique adopte une structure en bicouche lipidique.C. On retrouve du cholestérol dans les membranes plasmiques.D. Les glycolipides sont retrouvés dans le feuillet externe exclusivement.E. Des protéines hydrophobes peuvent diffuser librement dans la membrane plasmique.
II- Lipides saponifiables
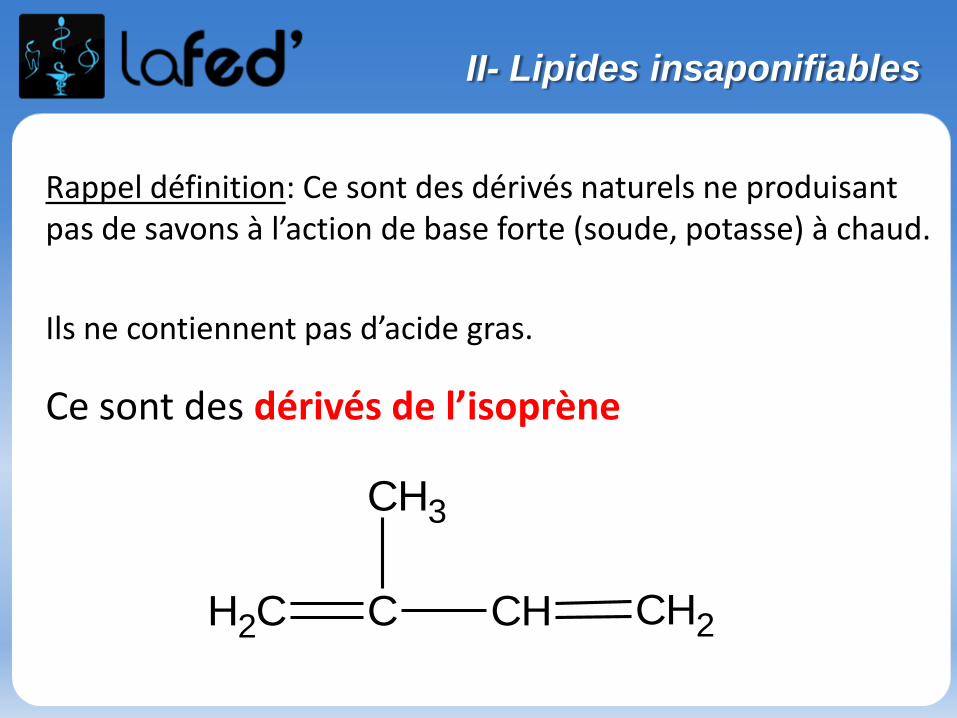
Rappel définition: Ce sont des dérivés naturels ne produisant pas de savons à l’action de base forte (soude, potasse) à chaud.
Ils ne contiennent pas d’acide gras.
Ce sont des dérivés de l’isoprène
CH2 C
CH3
CH CH2
II- Lipides insaponifiables
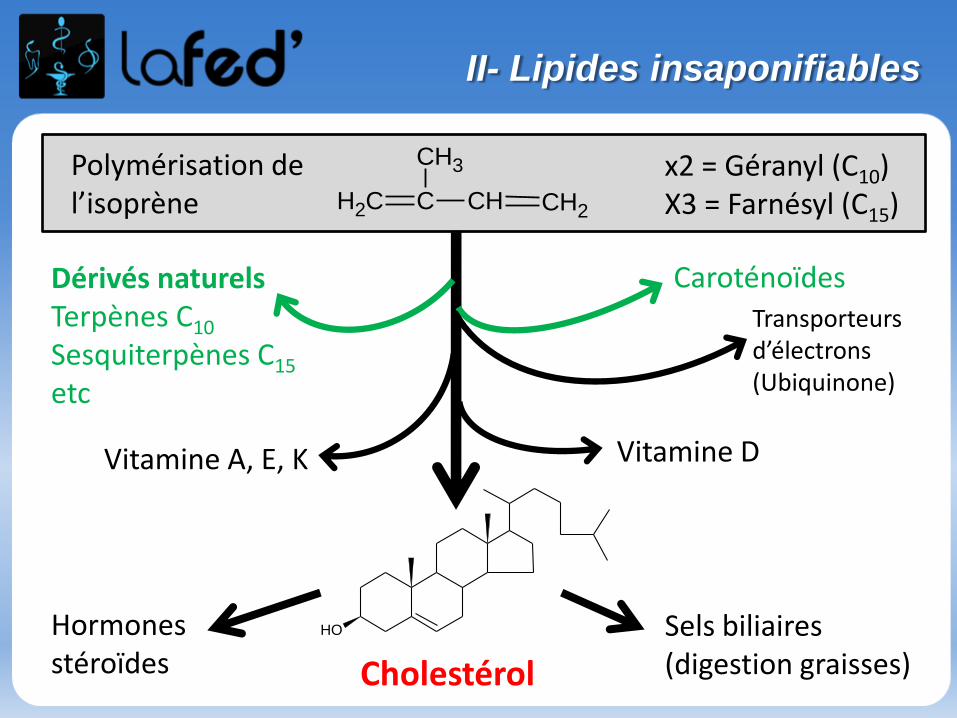
CH2 C
CH3
CH CH2
OH
Vitamine D
Hormones stéroïdes
Sels biliaires(digestion graisses)Cholestérol
Vitamine A, E, K
CaroténoïdesTransporteurs d’électrons(Ubiquinone)
Polymérisation de l’isoprène
Dérivés naturels Terpènes C10
Sesquiterpènes C15
etc
x2 = Géranyl (C10)X3 = Farnésyl (C15)
II- Lipides insaponifiables
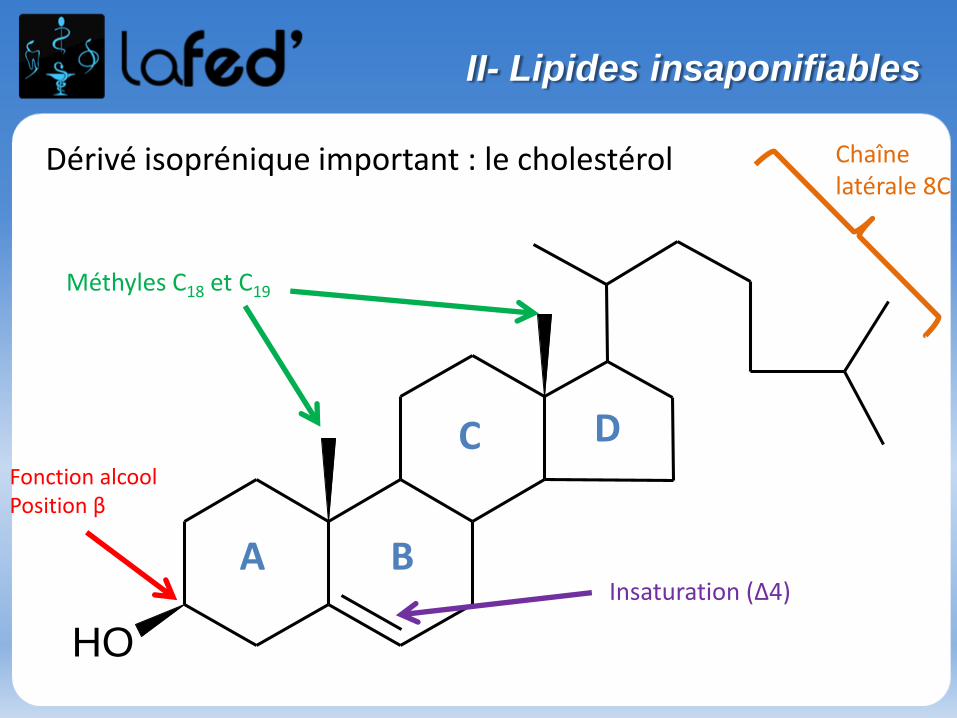
Dérivé isoprénique important : le cholestérol
OH
Fonction alcoolPosition β
A B
C D
Méthyles C18 et C19
Insaturation (Δ4)
Chaîne latérale 8C
II- Lipides insaponifiables
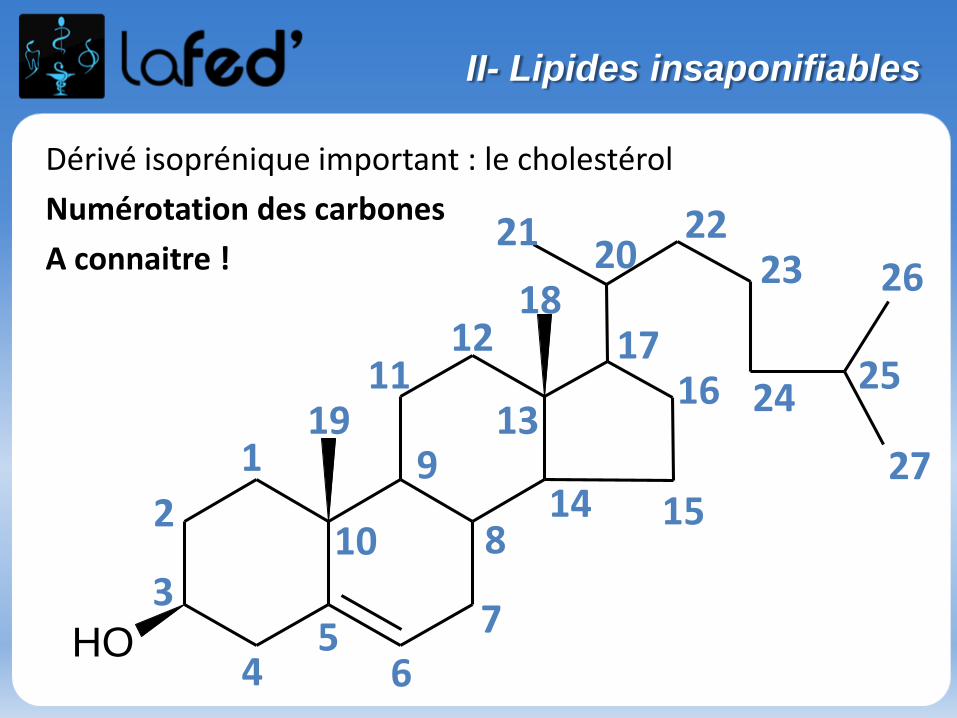
Dérivé isoprénique important : le cholestérol
Numérotation des carbones
A connaitre !
OH
1
2
3
45
6
7
8
9
10
1112
13
14 15
1617
18
19
2021 22
23
2425
26
27
II- Lipides insaponifiables
Exercice
Le cholestérol est tout nu, il faut le rhabiller !
II- Lipides insaponifiables
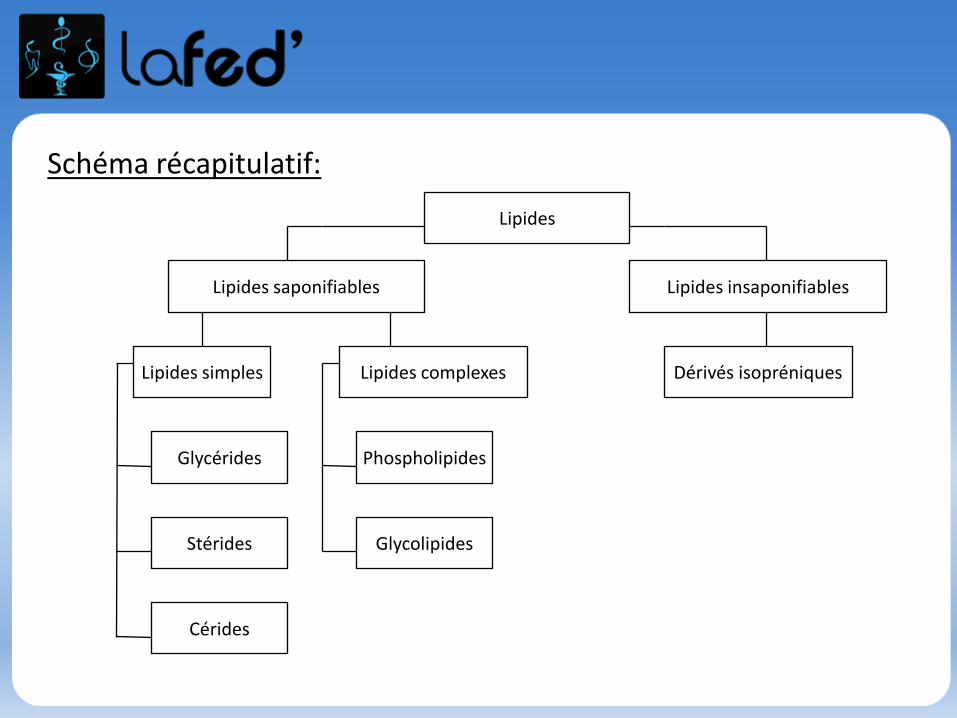
Schéma récapitulatif:
Lipides saponifiables Lipides insaponifiables
Lipides
Lipides simples Lipides complexes Dérivés isopréniques
Glycérides
Stérides
Cérides
Phospholipides
Glycolipides

Stage de Pré-Rentrée 2012UE 1 – Notions d’enzymologie
333
Diaporama réalisé par les tuteurs de La Fed’

I- 1 Généralités
Qu'est-ce qu'un ligand ?Molécule généralement plus petite qu'une protéine (ion, acide aminé, hormone,médicament)
Association avec la protéine généralement non covalente, saturable, réversible, spécifique sur une petite surface (10 à 30aa max) et en présence d'eau.
Exemples
enzyme-substrat, récepteur-hormone, récepteur-neurotransmetteur, récepteur-médicament
Rôles
transport (ion fer), thérapeutique, interactions intercellulaires...
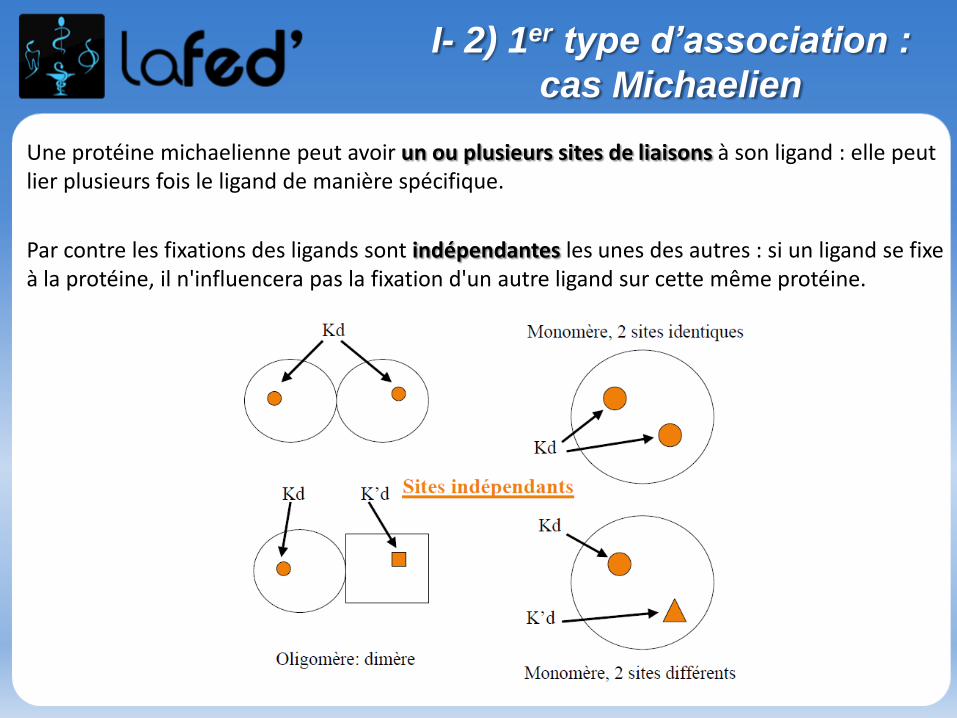
I- 2) 1er type d’association :
cas Michaelien
Une protéine michaelienne peut avoir un ou plusieurs sites de liaisons à son ligand : elle peut lier plusieurs fois le ligand de manière spécifique.
Par contre les fixations des ligands sont indépendantes les unes des autres : si un ligand se fixe à la protéine, il n'influencera pas la fixation d'un autre ligand sur cette même protéine.

De manière générale: vitesse = k x concentration réactif(s)
Pour la formation : vformation = k1 x P x L
Pour la dissociation : vdissociation = k-1 x PL
Et à l’équilibre: vformation = vdissociation donc k1 x P x L = k-1 x PL
k-1/k1 = (P x L) / PL = Kd (mol.L-1) = L0.5 = 1/Ka (L.mol-1)
I- 2) 1er type d’association :
cas Michaelien

Kd
• est une constante de dissociation, donc pour un équilibre donné elle ne varie qu'avec la température, et pas avec la concentration.
• indique la concentration en ligand pour laquelle la protéine est à moitié saturée . Ce n'est pas forcément à l'équilibre.
• plus elle augmente, moins la liaison avec le ligand est affine.
• se mesure avec la dialyse à l’équilibre (si un site) et avec le méthode de SCATCHARD (si plusieurs sites).
I- 2) 1er type d’association :
cas Michaelien
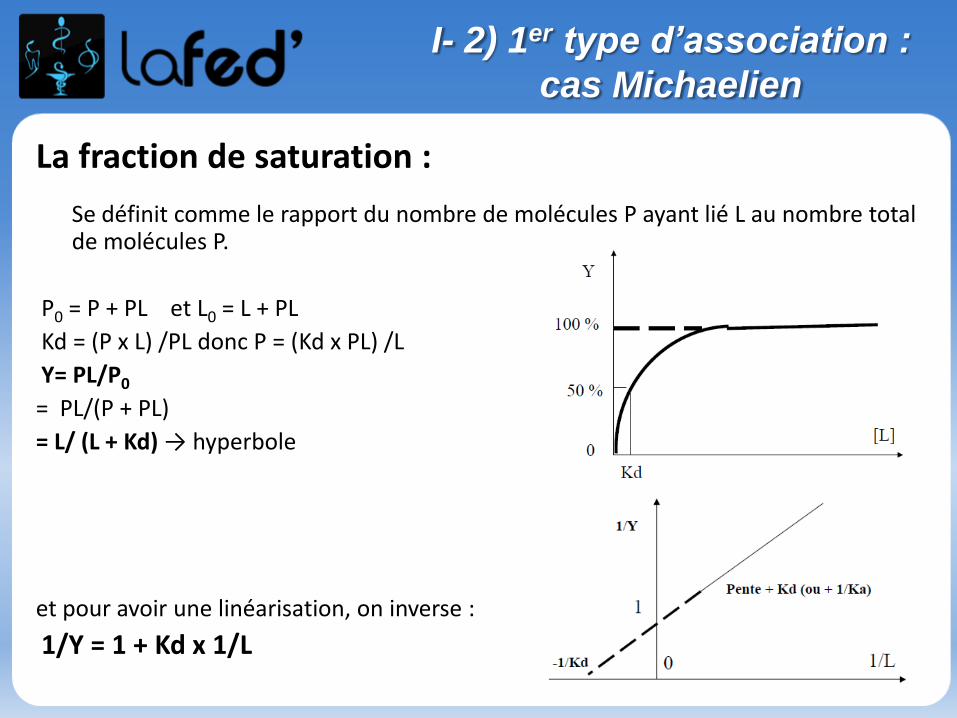
La fraction de saturation :
Se définit comme le rapport du nombre de molécules P ayant lié L au nombre total de molécules P.
P0 = P + PL et L0 = L + PL
Kd = (P x L) /PL donc P = (Kd x PL) /L
Y= PL/P0
= PL/(P + PL)
= L/ (L + Kd) → hyperbole
et pour avoir une linéarisation, on inverse :
1/Y = 1 + Kd x 1/L
I- 2) 1er type d’association :
cas Michaelien

L'allostérie se définit par le fait qu'une protéine a plusieurs sites identiques ou différents qui interagissent entre eux (structure quaternaire).
Cette dépendance des sites entre eux s'illustre par le nombre de Hill n. Le nombre de Hill est le nombre de sites en interaction.
• Si n=1 : cas michaelien (les sites sont indépendants, chaque site n'interagit qu'avec lui même donc n=1)
• Si n>1, coopération positive : la fixation du ligand facilite la fixation d'un autre ligand sur un autre site en augmentant son affinité.
• Si n<1, coopération négative : la fixation du ligand gène celle d'un autre sur un autre site en diminuant son affinité.
Remarque : n ne peut pas être ≤ 0.
I- 3) 2ème type d’association :
cas allostérique

La fraction de saturation devient alors:
• Le nombre de Hill intervient au niveau de la puissance.
• Ici, on ne parle pas de Kd, mais de L0,5 . Le Kd est une constante or ici L0,5 dépend de n, donc ce n'est plus une constante.
• Pour le cas michaelien, on a n = 1, donc Kd c'est donc L0,51.
I- 3) 2ème type d’association :
cas allostérique
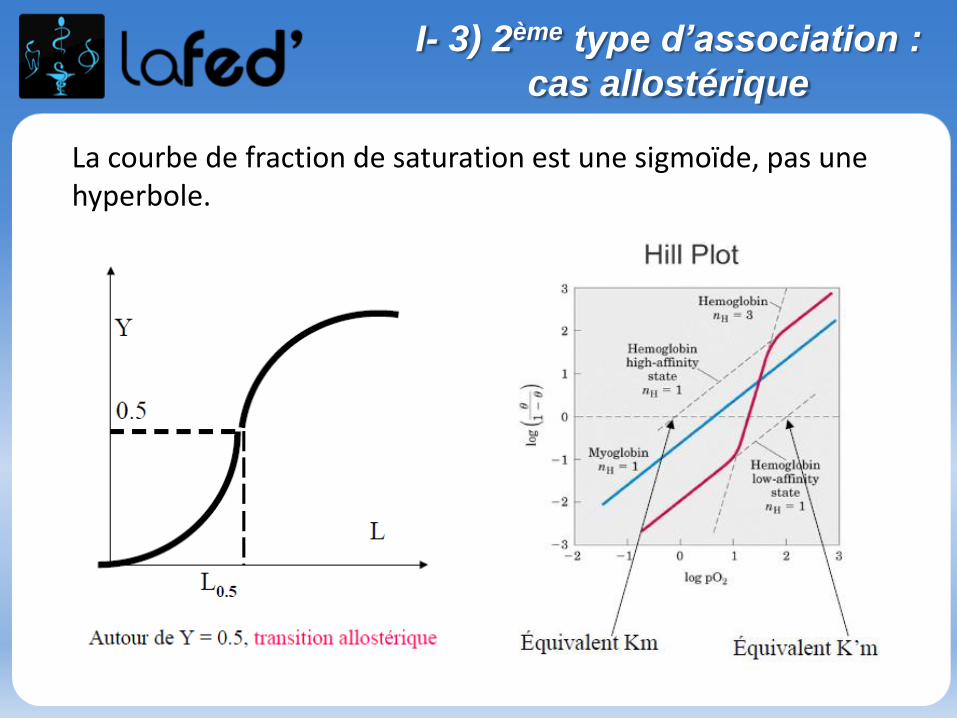
La courbe de fraction de saturation est une sigmoïde, pas une hyperbole.
I- 3) 2ème type d’association :
cas allostérique
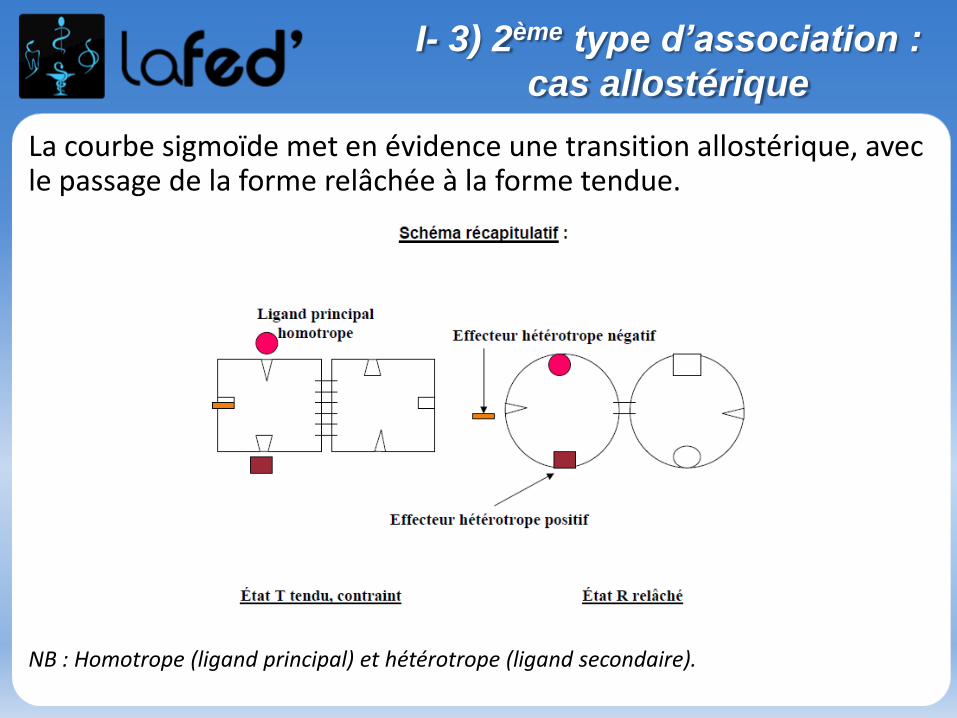
La courbe sigmoïde met en évidence une transition allostérique, avec le passage de la forme relâchée à la forme tendue.
NB : Homotrope (ligand principal) et hétérotrope (ligand secondaire).
I- 3) 2ème type d’association :
cas allostérique

II- La relation enzyme-substrat
Les enzymes:
• Une enzyme peut être une protéine mais également un ARN (ribozyme).
• Catalyseurs qui diminuent le DGa (donc augmente la vitesse de la réaction) mais qui ne permettent pas de déclencher la réaction (réactions sans enzymes possibles) donc ne modifient pas le ∆G.
• Ne vivent pas à l'infini (turn over).
• Ne sont pas modifiées en fin de réaction.
• Elles catalysent le plus souvent des réactions irréversibles in vivo (enzyme différente dans chacun des 2 sens d'une réaction).
• Une enzyme est caractérisée par des certains paramètres : affinité, vitesse et activité.

L’affinité enzymatique:
• Ks = k-1/k1 = E x S / ES ( Ks = Kd d'une enzyme).
• Km = E x S / ES = k-1+ k2/ k1 or k2 petit donc Km = Ks = Kd (en arrondissant).
II- La relation enzyme-substrat

La vitesse
• Par extension il existe une formule similaire à la saturation, c’est la vitesse.
• (v / Vm) = S / (S + Km) tout comme Y = L / (L + Kd), ce sera la même courbe hyperbolique.
• On passe d'une vitesse d'ordre 1 à une vitesse d'ordre 0.
• Km= concentration pour laquelle enzyme à moitié saturée et pour laquelle vitesse est à la moitié de son max.
Remarque : vitesse conditionnée par k2 car k2 est le plus petit (v = k2 x ES).
II- La relation enzyme-substrat
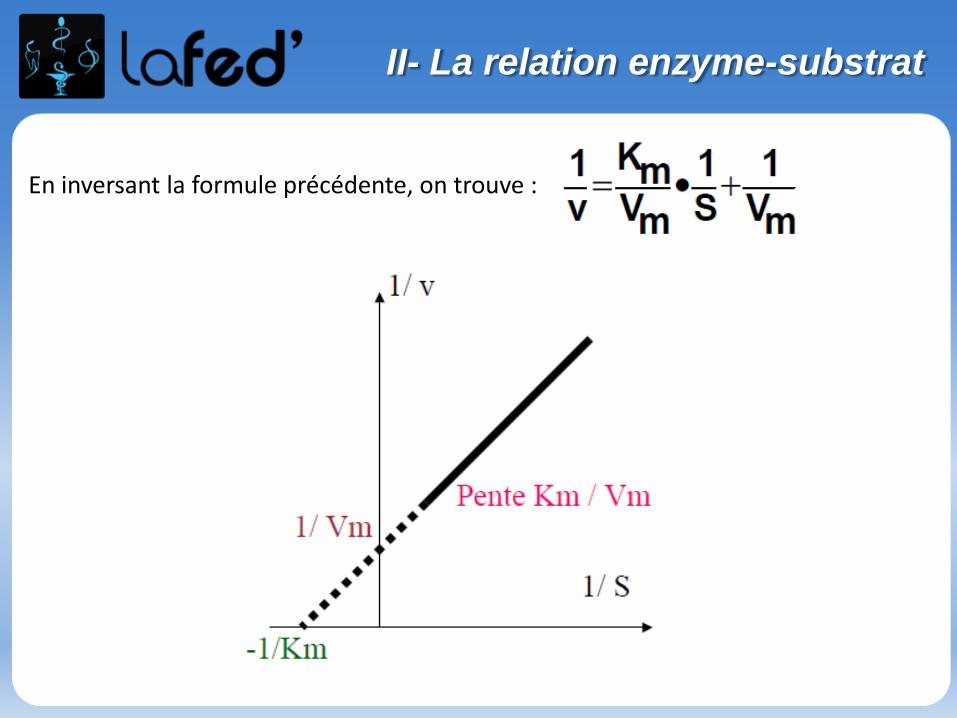
En inversant la formule précédente, on trouve :
II- La relation enzyme-substrat
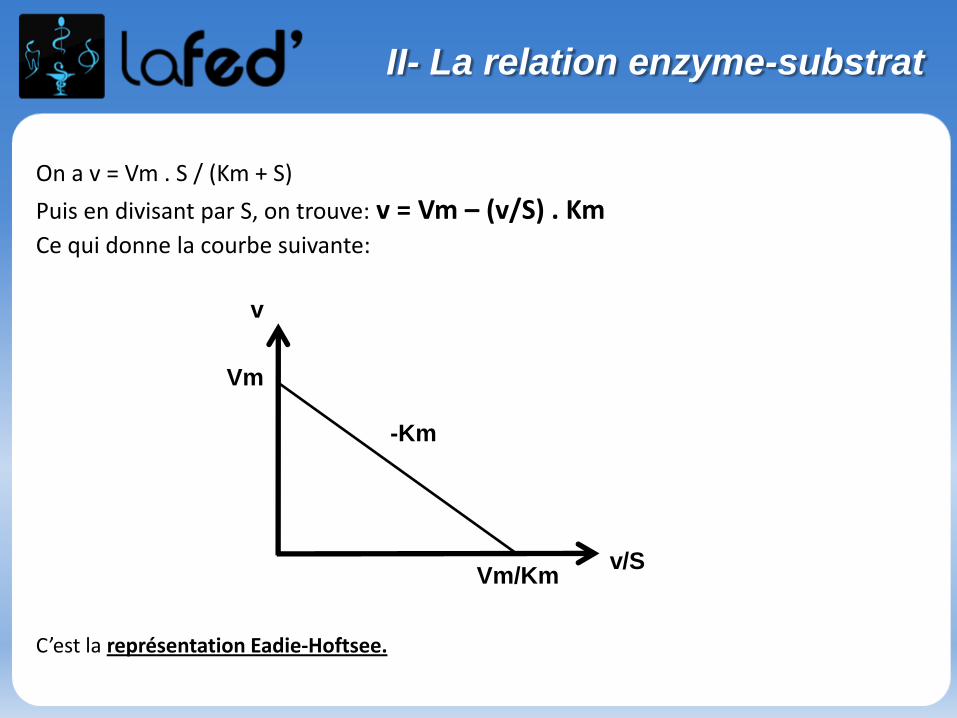
On a v = Vm . S / (Km + S)
Puis en divisant par S, on trouve: v = Vm – (v/S) . KmCe qui donne la courbe suivante:
C’est la représentation Eadie-Hoftsee.
II- La relation enzyme-substrat
Vm
Vm/Kmv/S
-Km
v

L’activité:
• Vitesse maximale Vm.
• Activité spécifique en UI = micromoles de substrat transformées par minute et par quantité d'enzyme (gramme, kg,...)
• Activité moléculaire = moles de substrat transformées par seconde et par mole d'enzyme, unité en s-1.
• k2 = Vm / E0 (activité moléculaire)
• Remarque : Le critère d'efficacité tient compte de l'affinité et de la vitesse, c'est k2/Km. Plus ce rapport est grand, plus l'enzyme est efficace car elle sera plus rapide (k2 plus grand) ou plus affine (Km plus petit). On tend vers la perfection cinétique.
II- La relation enzyme-substrat

III- Inhibitions
Irréversibles :
• Réactions chimiques (intoxications aux pesticides)
• pH
• température élevée
→ dénaturent l’enzyme.

Réversibles
Inhibition compétitive (IC) :
L’inhibiteur (IC) est souvent un analogue de substrat, de telle sorte qu’il se loge à la place de S, sans être en général transformé.
On a alors: v = Vm.S/(K’m +S) avec K’m = Km( 1+ I/Ki)
III- Inhibitions

Inhibition non compétitive (INC) :
L’inhibiteur se fixe en dehors du site actif. Il va freiner la cinétique sans toucher l’affinité.
III- Inhibitions
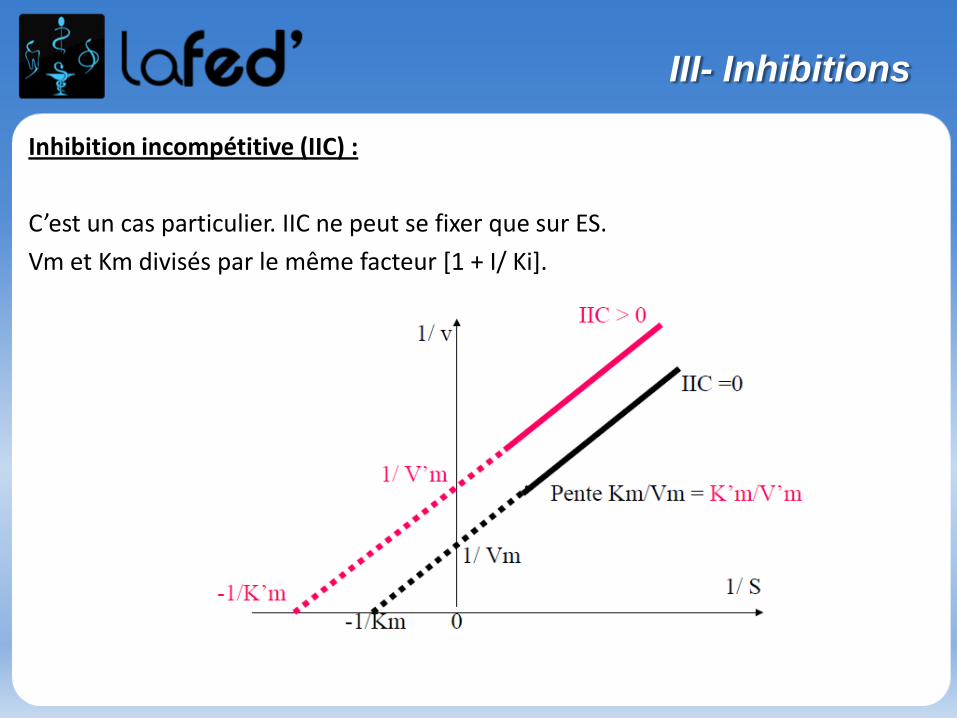
Inhibition incompétitive (IIC) :
C’est un cas particulier. IIC ne peut se fixer que sur ES.
Vm et Km divisés par le même facteur [1 + I/ Ki].
III- Inhibitions

Stage de Pré-Rentrée 2012UE 1 – Les acides nucléiques
353
Diaporama réalisé par les tuteurs de La Fed’

I- Introduction
Les acides nucléiques sont à la base du vivant. Parmi eux, on connait
notamment l’ADN, présent dans toutes les cellules nucléées de
l’organisme.
Un nucléotide est formé :
- d’une base azotée,
-d’un ose (sucre) qui déterminera le type d’acide nucléique,
- d’un acide phosphorique .
C’est l’enchainement des nucléotides qui forme un acide nucléique.
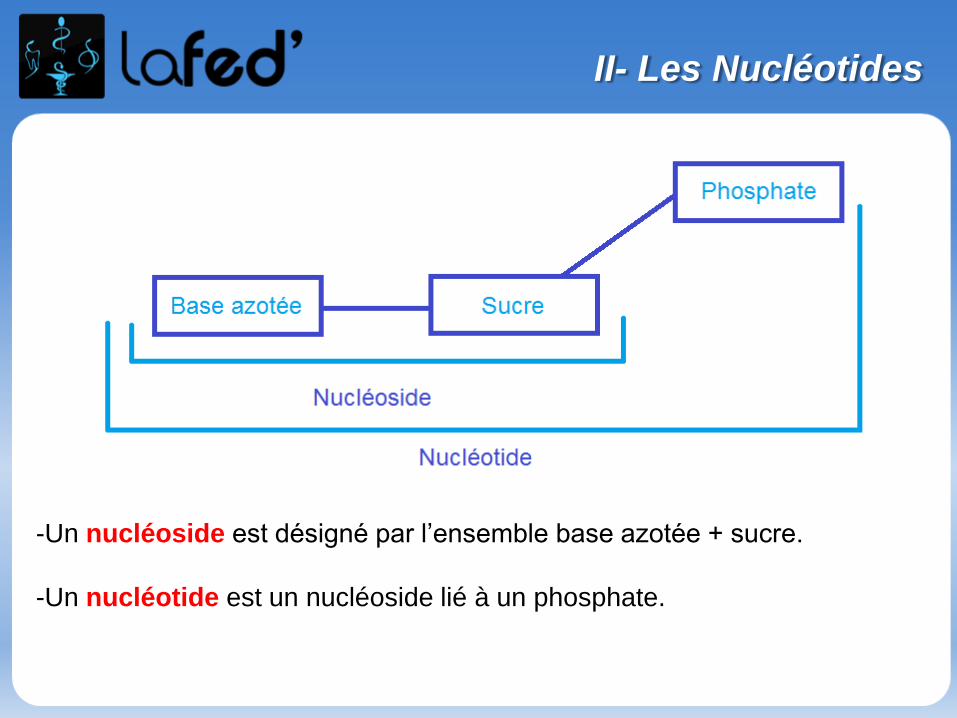
-Un nucléoside est désigné par l’ensemble base azotée + sucre.
-Un nucléotide est un nucléoside lié à un phosphate.
II- Les Nucléotides

Les bases azotées
Elles sont de deux types : les bases puriques et les bases pyrimidiques.
Les bases puriques :
Ce sont les bases formées à partir du noyau purine. Ce noyau est constitué de
deux hétérocycles aromatiques : un cycle pyrimidine et un cycle imidazole .
Voici sa représentation :
N
N
NH
N1
2
3
4
567
8
9
II- Les Nucléotides
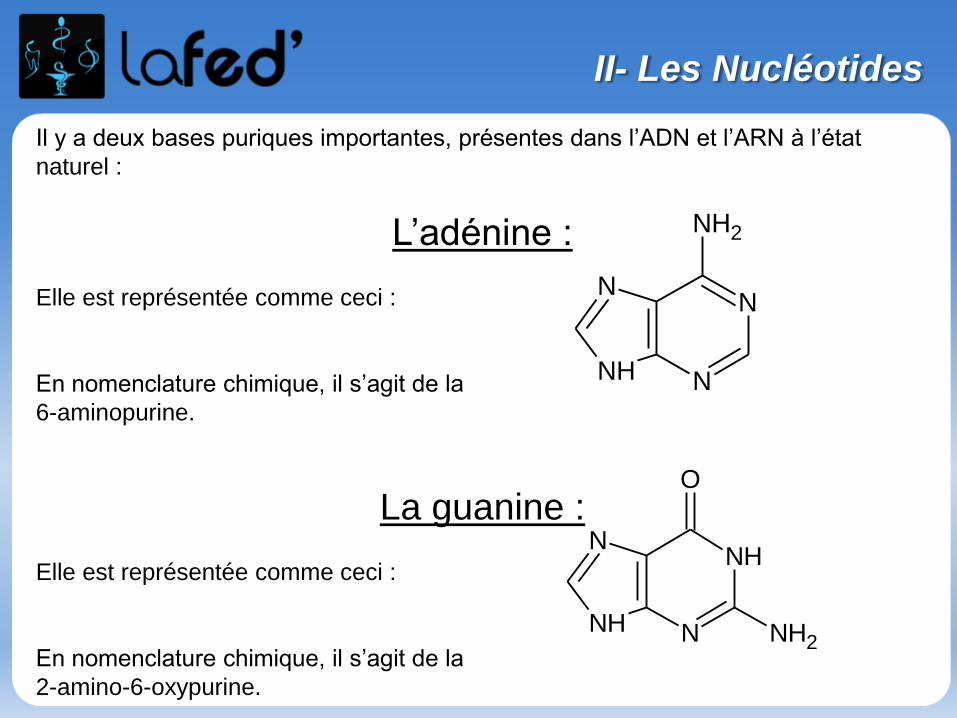
Il y a deux bases puriques importantes, présentes dans l’ADN et l’ARN à l’état
naturel :
L’adénine :
Elle est représentée comme ceci :
En nomenclature chimique, il s’agit de la
6-aminopurine.
La guanine :
Elle est représentée comme ceci :
En nomenclature chimique, il s’agit de la
2-amino-6-oxypurine.
N
N
NH
N
NH2
N
NH
NH
N
NH2
O
II- Les Nucléotides
Les bases pyrimidiques :
Ce sont les bases formées à partir du noyau pyrimidine. A la différence du noyau
purique, le noyau pyrimidique est formé d’un seul hétérocycle aromatique.
Voici sa représentation :
1
2
3
4
5
6
N
NH
La numérotation diffère entre les noyaux puriques et pyrimidiques !!!!
II- Les Nucléotides
Il y a trois bases pyrimidiques importantes :
La cytosine :
Elle est représentée comme ceci :
En nomenclature chimique, il s’agit de la
2-oxy-4-aminopyrimidine.
Elle est aussi bien présente au sein de l’ADN que de l’ARN à l’état naturel.
N
NH
NH2
O
II- Les Nucléotides
La thymine :
Elle est représentée comme ceci :
En nomenclature chimique, il s’agit de la
5-méthyl-2,4-dioxypyrimidine.
Attention : elle est bien présente dans l’ADN à l’état normal. Cependant, elle est
absente de tout ARN naturel !! Dans les ARN, la thymine sera remplacée par l’uracile.
L’uracile :
Elle est représentée comme ceci :
En nomenclature chimique, il s’agit de la
2,4-dioxypyrimidine.
Elle remplace la thymine au sein des ARN, on en retrouve normalement pas dans
l’ADN.
NH
NH
O
O
CH3
NH
NH
O
O
II- Les Nucléotides
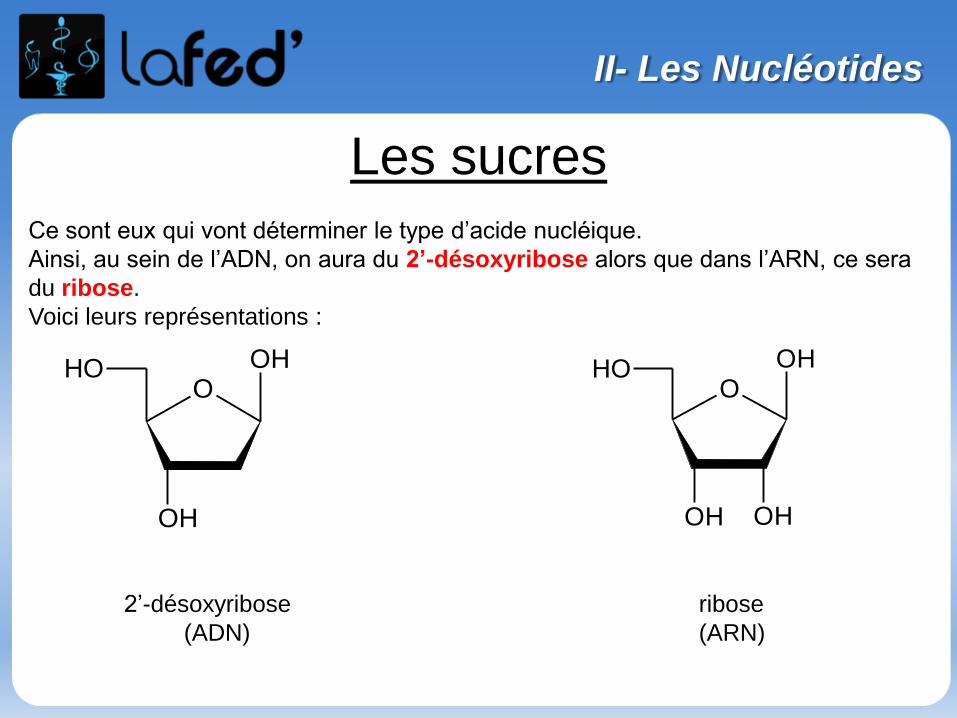
Les sucres
Ce sont eux qui vont déterminer le type d’acide nucléique.
Ainsi, au sein de l’ADN, on aura du 2’-désoxyribose alors que dans l’ARN, ce sera
du ribose.
Voici leurs représentations :
2’-désoxyribose ribose
(ADN) (ARN)
OHO
OH
OH
OHO
OH
OHOH
II- Les Nucléotides

-Ces oses sont, au sein des acides nucléiques, sous la forme furanose.
Petit rappel sur le cours des glucides : la forme furanose est la forme cyclique des
oses formée par réaction d’hémiacétalisation entre le carbonyle et la fonction alcool
du carbone 4. Il s’agit d’un cycle à 5 sommets.
- Dans les acides nucléiques, les oses ont la configuration L.
- Ils sont en conformation b (le OH anomérique et le C5 se trouvent du même côté
du plan).
II- Les Nucléotides

Le phosphate :
L’acide phosphorique H3PO4 peut être représenté comme ceci :
Il possède donc 3 fonctions acides. Au sein des nucléotides, une de ces fonctions
va s’estérifier avec la fonction alcool libre en C5’ du sucre.
Les nucléotides à l’état libre peuvent être mono, di ou triphosphates. La liaison entre
ces phosphates est une liaison anhydride d’acide. Cependant dans les acides
nucléiques, ils sont monophosphates.
P
O
OH
OH
OH
II- Les Nucléotides
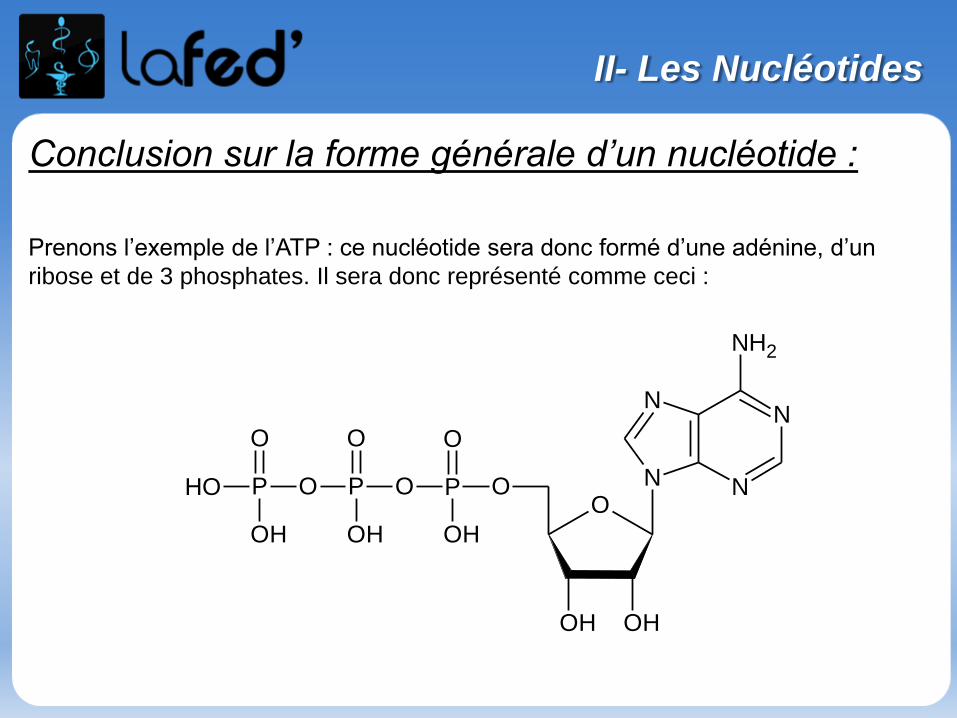
Conclusion sur la forme générale d’un nucléotide :
Prenons l’exemple de l’ATP : ce nucléotide sera donc formé d’une adénine, d’un
ribose et de 3 phosphates. Il sera donc représenté comme ceci :
II- Les Nucléotides
NO
O
OHOH
P
O
OH
P
O
O
OH
OH
OH
O
O
P N
NN
NH2
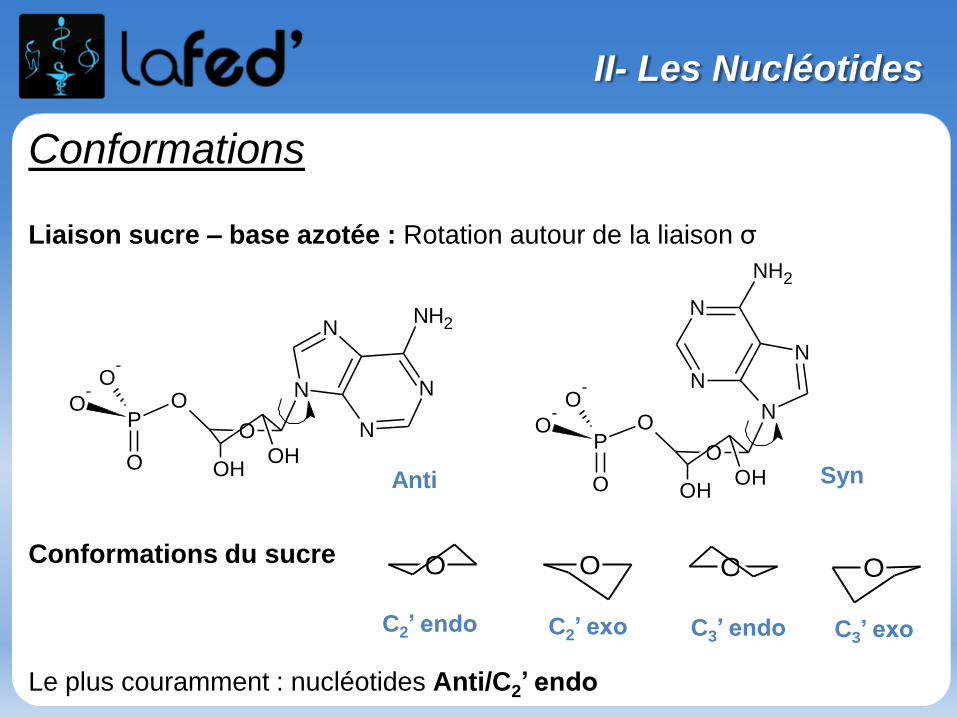
Conformations
Liaison sucre – base azotée : Rotation autour de la liaison σ
Conformations du sucre
Le plus couramment : nucléotides Anti/C2’ endo
II- Les Nucléotides
O
N
N
N
NNH2
OP
O-
O
O-
OHOH
SynAnti
O
NO
PO
-
O
O-
OHOH
N
N
N
NH2
O O O O
C2’ endo C2’ exo C3’ endo C3’ exo

Absorbance :
- Les acides nucléiques absorbent la lumière dans l’UV.
- On observe que les bases puriques absorbent mieux que les bases pyrimidiques.
- Le maximum d’absorbance est pour une longueur d’onde de 260 nm. Ainsi, on
utilise le rapport des absorbances suivant pour déterminer la pureté d’un acide
nucléique :
260nm (ac. nucléiques)/ 280nm (protéines)
.
II- Les Nucléotides
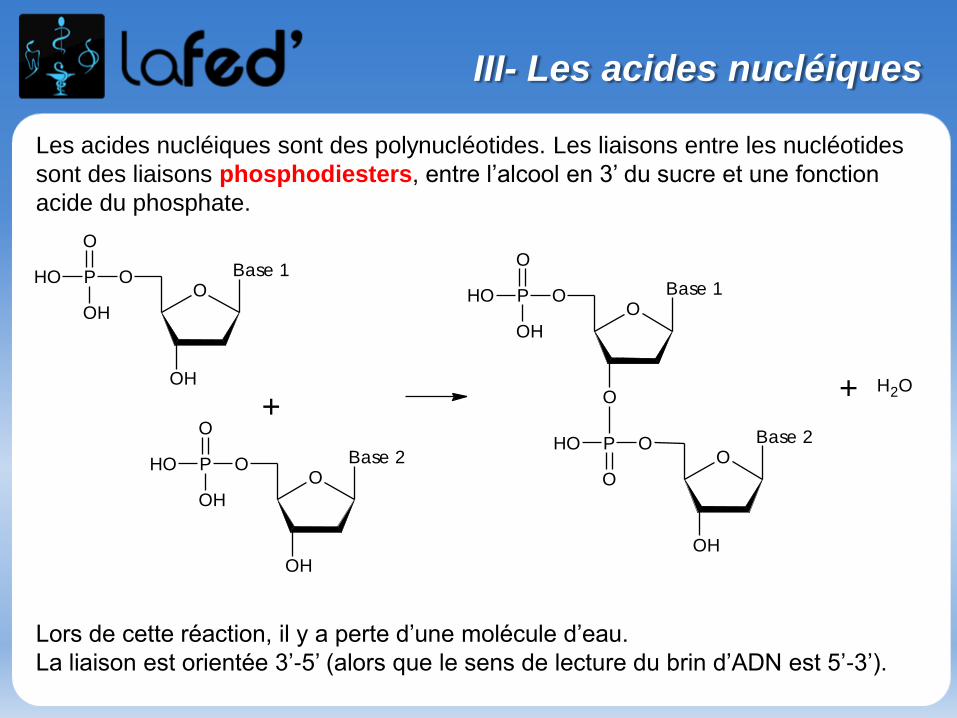
III- Les acides nucléiques
Les acides nucléiques sont des polynucléotides. Les liaisons entre les nucléotides
sont des liaisons phosphodiesters, entre l’alcool en 3’ du sucre et une fonction
acide du phosphate.
Lors de cette réaction, il y a perte d’une molécule d’eau.
La liaison est orientée 3’-5’ (alors que le sens de lecture du brin d’ADN est 5’-3’).
Base 1O
O
OH
OH
OH
O
P
+
Base 2O
O
OH
OH
OH
O
P
Base 2O
OH
Base 1O
O
O
OH
OH
O
P
P O
O
OH
+ H2O
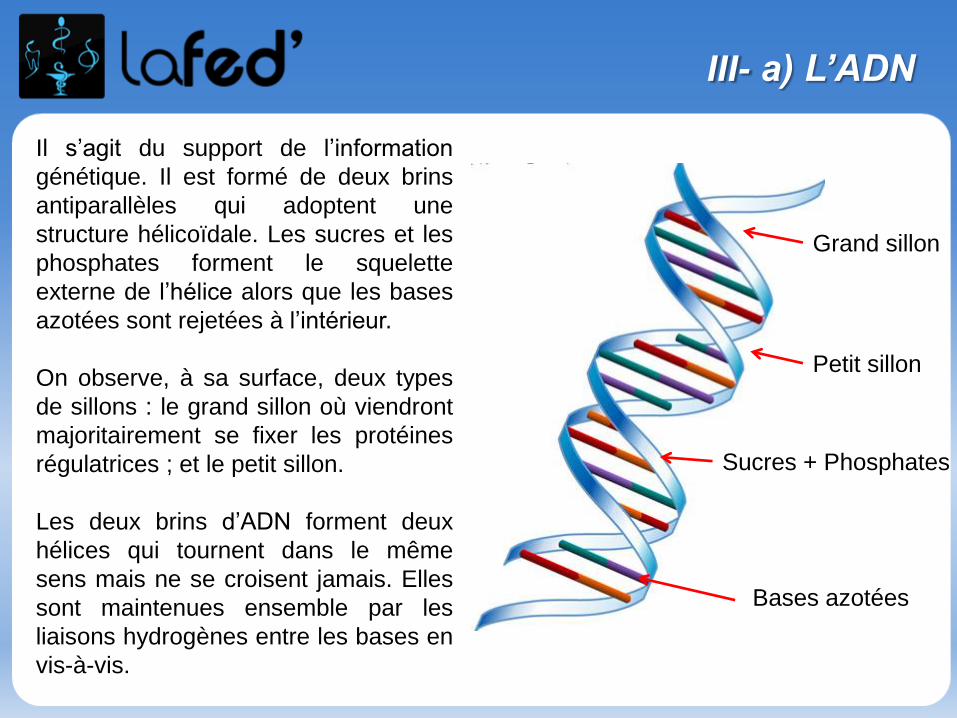
III- a) L’ADN
Il s’agit du support de l’information
génétique. Il est formé de deux brins
antiparallèles qui adoptent une
structure hélicoïdale. Les sucres et les
phosphates forment le squelette
externe de l’hélice alors que les bases
azotées sont rejetées à l’intérieur.
On observe, à sa surface, deux types
de sillons : le grand sillon où viendront
majoritairement se fixer les protéines
régulatrices ; et le petit sillon.
Les deux brins d’ADN forment deux
hélices qui tournent dans le même
sens mais ne se croisent jamais. Elles
sont maintenues ensemble par les
liaisons hydrogènes entre les bases en
vis-à-vis.
Grand sillon
Petit sillon
Sucres + Phosphates
Bases azotées
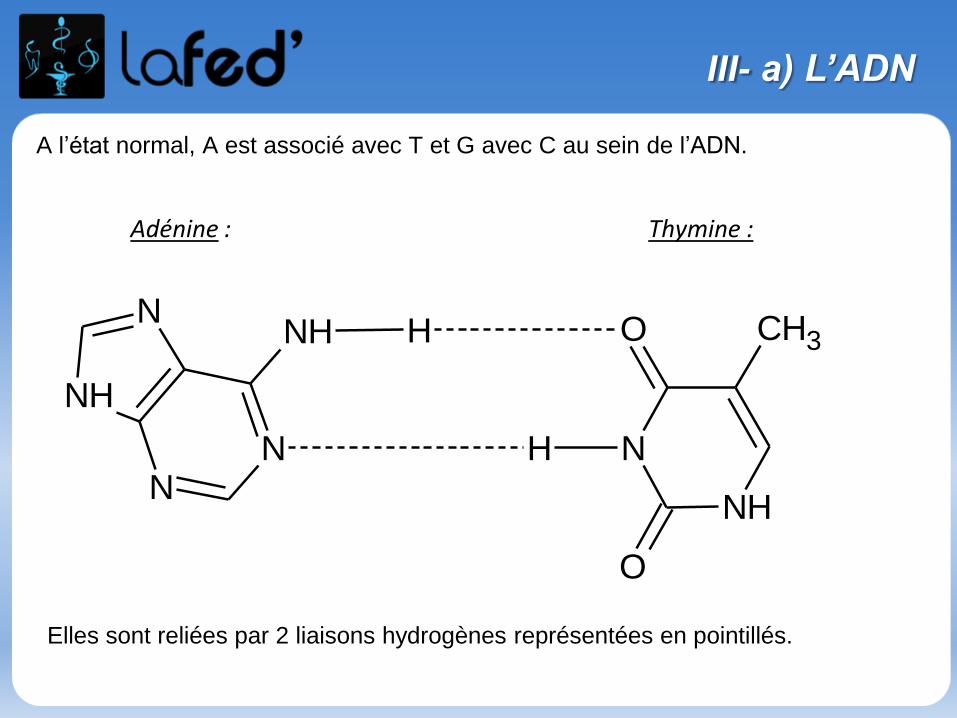
A l’état normal, A est associé avec T et G avec C au sein de l’ADN.
Adénine : Thymine :
NN
NH
NNH H
NH
N
O
O
CH3
H
III- a) L’ADN
Elles sont reliées par 2 liaisons hydrogènes représentées en pointillés.
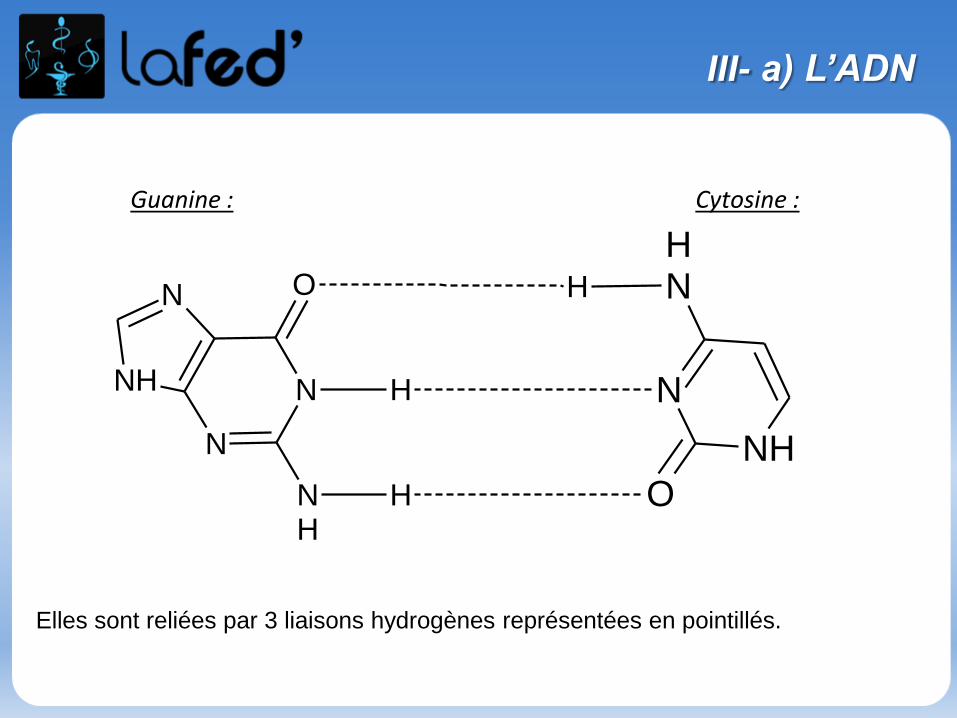
Guanine : Cytosine :
Elles sont reliées par 3 liaisons hydrogènes représentées en pointillés.
N
NNH
N
NH
O
H
H
N
NH
NH
O
H
III- a) L’ADN

III- a) L’ADN
Forme B de l’ADN :
- Il s’agit de la forme naturelle dans les conditions physiologiques.
- C’est une hélice de pas droit dont le plan des bases est perpendiculaire à l’axe de
l’hélice.
Elle comporte deux sillons, l’un majeur et l’autre mineur. Par tour d’hélice (34 Å), il y
a 10 paires de nucléotides.
Anti/C2’ endo

Forme A de l’ADN :
- Elle est observée dans certaines régions d’ADN naturel en milieu déshydraté,
lorsqu’il y a une forte concentration en cations.
- C’est une hélice de pas droit dont le plan des bases est légèrement incliné par
rapport à celui de l’hélice B.
Le sillon étroit est profond et inaccessible alors que l’autre est large et superficiel.
Par tour d’hélice (26 à 28Å), il y a 11 paires de nucléotides.
Anti/C3’ endo
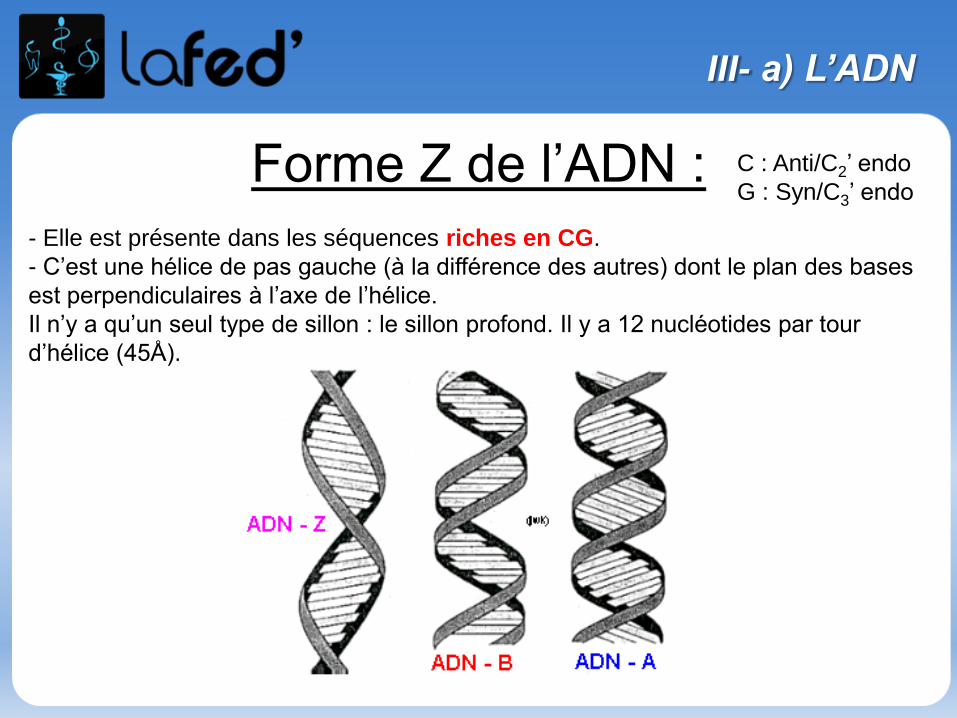
III- a) L’ADN
Forme Z de l’ADN :
- Elle est présente dans les séquences riches en CG.
- C’est une hélice de pas gauche (à la différence des autres) dont le plan des bases
est perpendiculaires à l’axe de l’hélice.
Il n’y a qu’un seul type de sillon : le sillon profond. Il y a 12 nucléotides par tour
d’hélice (45Å).
C : Anti/C2’ endo
G : Syn/C3’ endo

III- b) L’ARN
Petits rappels :
- Dans l’ARN, le sucre n’est pas le désoxyribose mais le ribose.
- La base T est remplacée par le U.
- Généralement, les ARN, à la différence de l’ADN, sont simples brins.
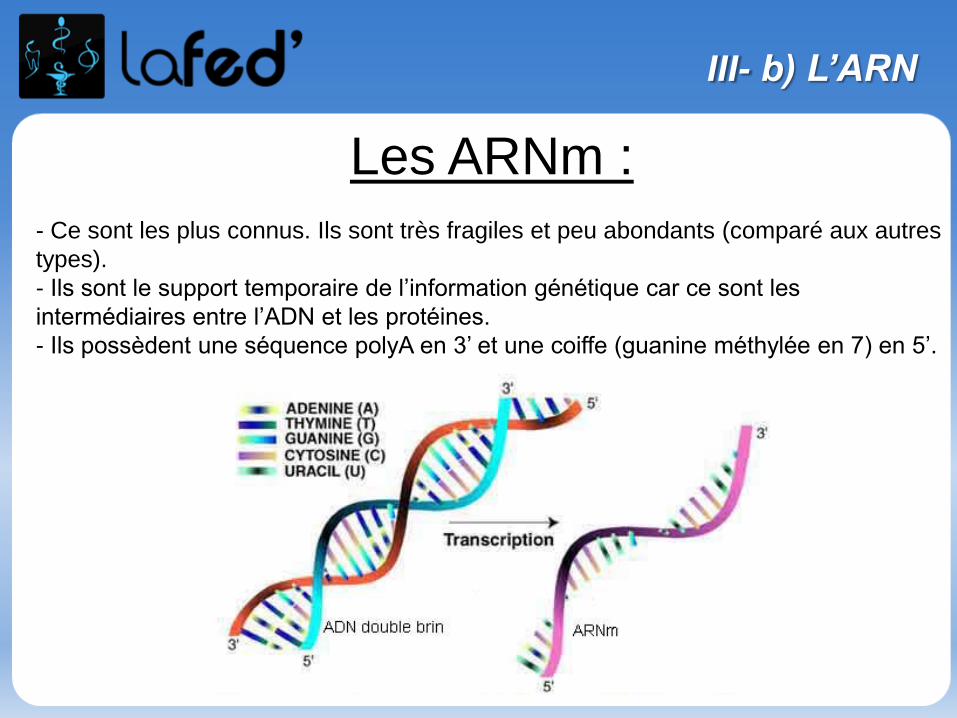
III- b) L’ARN
Les ARNm :
- Ce sont les plus connus. Ils sont très fragiles et peu abondants (comparé aux autres
types).
- Ils sont le support temporaire de l’information génétique car ce sont les
intermédiaires entre l’ADN et les protéines.
- Ils possèdent une séquence polyA en 3’ et une coiffe (guanine méthylée en 7) en 5’.
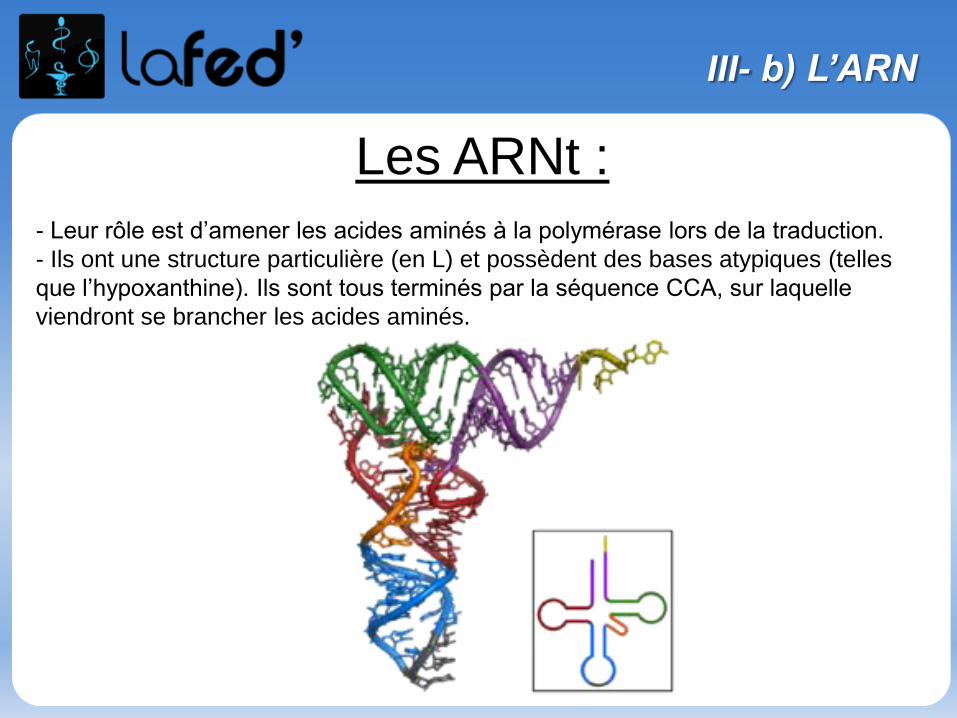
Les ARNt :
- Leur rôle est d’amener les acides aminés à la polymérase lors de la traduction.
- Ils ont une structure particulière (en L) et possèdent des bases atypiques (telles
que l’hypoxanthine). Ils sont tous terminés par la séquence CCA, sur laquelle
viendront se brancher les acides aminés.
III- b) L’ARN

III- b) L’ARN
Les ARNr :
- Ce sont les plus nombreux. Ils servent de catalyseurs pour la synthèse des
protéines.
- Ils sont assemblés avec des protéines en plusieurs sous unités qui forment le
ribosome, présent notamment à la surface du réticulum endoplasmique rugueux.