Embed Size (px)
Citation preview

UE 2 – BiopathologieM. Capellen
Date : 08/11/2017 Plage horaire : 16h15 – 18h15
Promo : P2 2017 – 2018 Enseignant : M. Capellen (cours assuré par Merlio)
Ronéistes : Clara LAVOCAT / Manuela LAU FAT
TD2 Patho tumeurs
I. Objectif et rappel
II. Cancer du colon
III. Cancer mammaire 1. Histoire naturelle du carcinome infiltrant du sein. 2. Les facteurs pronostiques et les facteurs prédictifs dans le cancer du sein. 3. Types histologiques. 4. Facteurs pronostiques dans le cancer du sein. 5. Facteurs histologiques prédictifs dans le cancer du sein.
IV. Cancer pulmonaire 1. Observation clinique. 2. Examen histologique

I. Objectif : Donner des exemples de tumeurs nécessitant une intégration de données clinique, d’imagerie-radiologie, de pathologique et de génétique somatique pour le diagnostic et /ou la prise en charge thérapeutique.
Rappel :
(pas présenté cette année)
Quand on fait de la génétique somatique on s'intéresse à la modification des gènes spécifiques de la tumeur.
Ces 3 types de cancers vont nous montrer comment des techniques génétiques (séquençage de l'ADN, PCR, exploration chromosomique..) permettent de donner une thérapie ciblée selon le type de cancer (prostate, côlon...).
II. Cancer du colon
Intérêts du dossier :
- Illustrer la progression tumorale et sa mise en évidence par le bilan d’extension
- Illustrer l’utilisation du système TNM et rappeler ses principes principaux (évolution/mise à jour ; cTNM/ pTNM/ stade)
- CCR = cancer colorectal
- Montrer l'intérêt de l'analyse moléculaire pour le diagnostic de certains cancers héréditaires ou sporadiques
QCM 1 : Un homme de 45 ans qui n'a pas d’AEG, a du sang rouge mélangé aux selles depuis un mois (rectorragies). Ses antécédents montrent qu'il a eu plusieurs poussées hémorroïdaires (= varices au niveau anal) avec rectorragies éclaboussant la cuvette des toilettes. Que faire en 1ère intention ?
A- Test d’hémocultureB- ColoscopieC- FOGD (fibro oeso-gastro-duodénale)D- Toucher rectalE- Scanner TAP (Thorax Abdomen Pelvis) F- Sclérose des hémorroïdes
Bonnes réponses : B et D Mauvaises réponses : ACEF

A : la sensibilité n'est que de 50 % (=la présence de sang n’est pas détectée dans tous les cas), Il est réservé au dépistage organisé.Le test d’hémoculture permet de révéler la présence de sang de manière cryptique, celui qui n’est pas visible directement ; ici on sait qu’on a du sang de manière objective, donc on n’est pas obligés de faire ce test.Question élève : Est ce qu'on peut faire plusieurs tests pour augmenter la sensibilité ?Réponse prof : Ça pourrait en effet augmenter la probabilité, et puis maintenant il y a d'autres tests basés sur des anticorps qui permettent de reconnaître des marqueurs de cellules tumorales. Dans un futur prochain vont même arriver des tests génétiques qui vont détecter des mutations dans les cellules tumorales relâchées dans les selles.Intérêt test hémoccult : simple, pas cher, et applicable sur l'ensemble de la population.Inconvénient : problème également de spécificité car les hémorroïdes faussent le test, or ils sont fréquents.
C : le sang est rouge donc non digéré, donc postérieur à l’estomac, au processus de digestion, doncinutile
E : pour les bilans d’extensions. À ce stade, on ne connaît même pas le type de lésions.
F : à partir du moment où on voit des hémorragies comme ça, on ne va pas se contenter de scléroser les hémorroïdes, il y a certainement des lésions plus importantes à rechercher
B : pour voir si la lésion est plus en amont ; toute rectorragie = coloscopieSi TR négatif elle doit être pratiquée car elle permettrait de dépister des polypes avant qu'on fasse un cancer colorectal. Si ATCD de cancer colorectal dans la famille il faut encourager la personne a faire une coloscopie car ce cancer a des prédispositions familiales.
D : pour voir si la lésion est directement accessible. C'est le 1er examen que doit faire le médecin généraliste pour ce type de patient avant une consultation avec un spécialiste. «Le problème du toucher rectal c'est d'avoir les doigts assez longs» (trololol) sinon on va détecter que les tumeurs basses...
- Résultats des tests
dans la paroi du rectum, accessible au toucher
rectal + ici en coloscopie
Si la lésion était petite (polype) le médecin aurait pu enlever le pied de la tumeur : on aurait fait une biopsie exérèse (pince à lasso qui étrangle tumeur, en brûlant), d'où l'importance du diagnostic précoce.
Ici elle est trop grosse on peut pas l'enlever donc on fait juste une biopsie pour connaître la nature de la tumeur (ici il se doute que ça va être une tumeur épithéliale).

Ici on a à faire à un adénocarcinome car ça forme des glandes : cellules épithéliales en travées carcinomateuses qui infiltrent le chorion muqueux. C'est donc un adénocarcinome infiltrant avec des cellules carcinomateuses qui se détachent.
Juste avec un HES quand la tumeur est bien différenciée on a pas forcément besoin au départ d'HIC pour faire le diagnostic .
Si on était dans un ganglion on aurait pu se demander d'où vient cet adénocarcinome mais là on est sur le site primitif de la tumeur donc on raisonne pas de la même façon : on n'est pas dans une situation métastatique où on se demande où est le cancer primitif, là on est déjà sur le site du cancer primitif.
Ex biopsie tumeur cutanée : en général ce sont des tumeurs primitives (métastases au niveau cutané plus rare)
Les dysplasies légères ou sévères, sont des lésions intra-épithéliales. L’aspect d’une dysplasie varie selon le type de l’épithélium. Si on a un épithélium pavimenteux, il présente déjà plusieurs couches, ce qu’on appellera dysplasie correspondra alors à une désorganisation des couches. Par exemple, on ne retrouvera pas la couche basale, granuleuse, cornée etc, on aura l’impression d’avoir les mêmes cellules partout.
Lorsqu’on a affaire à un épithélium unistratifié, si on voit un aspect de stratification, c’est anormal.
Ici, on se trouve au niveau du rectum, on devrait avoir un épithélium de type intestinal avec des entérocytes et cellules caliciformes+++. On voit cependant de nombreux entérocytes avec une dysplasie assez sévère. Si l’on n’avait que ça, on dirait que c’est une dysplasie, parce qu’on a une désorganisation de l’épithélium, plus la même proportion de cellules, etc.
Cela dit, on voit également des petits boyaux de cellules épithéliales qui sont dans le chorion, à priori elles ne sont pas entourées de lame basale. Si l’on faisait des immunomarquages ou colorations spéciales, on verrait qu’il n’y a pas de LB autour. Ce sont donc des foyers d’infiltration de cellules épithéliales dans le chorion. Même sur les différentes incidences de coupe, des fois on peut voir des glandes de Lieberkhun etc, mais elles seront bien organisées. Là on voit des boyaux cellulaires qui n’ont rien à voir avec des glandes de Lieberkhun. Si on vérifie, il n’y a pas de LB autour, donc c’est un adénocarcinome.
Si les cellules épithéliales ont passé la lame basale, c’est un cancer infiltrant, invasif.
Question d’une élève : Pourquoi ne coche-t-on pas la A : dysplasie ?
Prof : c’est un QCM à choix unique ici, on précisera. Sinon on pourrait cocher les deux.
Quand on fait le diagnostic, on aurait pu cocher les deux, mais ce qui l’emporte dans le diagnostic, c’est la lésion la plus importante. Si le patient n’a que de la dysplasie, ok parce que ça peut être réversible. S’il a dysplasie + adénocarcinome infiltrant, c’est ce qui pèse le plus lourd dans le pronostic qui va entrer en compte dans le diagnostic. (attention diagnostic : dans la limite du prélèvement analysé)
Si les anapaths ont des doutes, ils peuvent vérifier avec une IHC, coloration spéciale (PAS, Alcian…), révélant qu’il n’y a pas de LB autour. Ces cellules ne sont pas arrivées là par le Saint-Esprit, elles proviennent donc de l’épithélium de départ, duquel elles se sont détachées pour infiltrer le chorion.
- Diagnostic : Adénocarcinome colo-rectal
- QCM 2 : Ses biopsies coliques révèlent un adénocarcinome rectal. Que préconisez-vous ?
A- une radiothérapie préopératoire pour faciliter la résécabilité. B- résection colique d'emblée.C- une surveillance.D- un bilan d'extension.

Bonne réponse : D : cela permet une évaluation de l'extension loco-régionale mais aussi à distance
Il faut savoir si on est face à une maladie localisée ( = intraépithéliale), localement avancée ( = infiltrante)
ou généralisée ( = métastatique).
Ces 3 termes sont importants en cancérologie et permettent de faire le pronostic de la maladie et de décider
du traitement (local limité, local important ou traitement généralisé comme la chimio ou per os).
Le bilan d'extension se fait à 2 niveaux : local et général.
Mauvaises réponses : A : Il n’y a visiblement pas de problème de résection. On ne sait pas s’il y a des
lésions ailleurs. Elle sera indiquée si l'on a une extension locale importante.
B : peut-être que la lésion est très limitée au rectum, du coup on ne va pas lui enlever son côlon, vu les conséquences que ça a. Elle sera indiquée si l'on a une extension locale peu importante.
C : on ne va pas faire juste une surveillance, car ça peut être bien plus grave.
on va donc faire un bilan d’extension (loco-régional + à distance)
- 1er bilan d’extension par écho endoscopie :
On a donc effectué ici une écho-endoscopie pour voir jusqu'où va l'invasion tumorale.
On observe la paroi musculeuse et on arrive presque à voir les deux plans musculaires : la musculeuse interne = circulaire interne et la musculeuse externe = longitudinale externe.
Il y a la muqueuse, puis 2 couches de musculeuse, puis l’adventice.
Sur une coupe transversale, on voit les fibres en longitudinal, c’est qu’elles tournent en rond, donc ce sont les circulaires. Si on les voit transversales, c’est qu’elles longent le tuyau, donc elles sont longitudinales.
Il n’y a pas de ganglion suspect sur cette image, donc on n’ira pas forcément plus loin en terme d’imagerie. On ne va pas forcément faire un scanner abdominal, une IRM etc.
Du point de vue ultrasonographique qui est égal à clinique, (Tumeur Node Métastase) :
- c = quand c’est par des examens cliniques d’imagerie, de palpation etc,
- p = pathological, c’est de l’histologie
Différents degrés d’envahissement :
TA ou CIS (carcinome in situ) : tumeurs intra-épithéliales
PT1 : envahit lame basale et chorion +/- profond ( PT1a, PT1b)
T2 : envahit le muscle (pour les épithéliums de revêtement en tout cas)
T3,T4… : envahit organes +/- éloignés de la tumeur initiale
Puis les M et N s’y rajoutent.
ici c’est N0, mais attention toujours dans la limite de ce qu’on a pu examiner
il peut peut-être y avoir du N1 et du M1 à distance.

On a plus d’information que la coloscopie car ici on a aussi une idée de ce que se passe en profondeur et pas seulement une vision de la surface.
Les cellules musculaires sont substitués par les cellules épithéliales tumorales qui sont moins denses que les fibres musculaires d’où cet aspect hypodense (noir) au niveau de la lésion tumorale.
Ceci est une vue macroscopique, il n’est pas sûr à 100% que la musculeuse externe est respectée du point de vue macroscopique. Il va donc y’avoir un geste chirurgical, une pièce opératoire qui va être analysé en histologie. On est un stade clinique cTNM puis on avoir recours a des techniques microscopiques pour faire le stade pTNM.
Ici on est au stade clinique T2 car la tumeur envahit la musculeuse et N0 car on ne voit pas de nodules suspects.
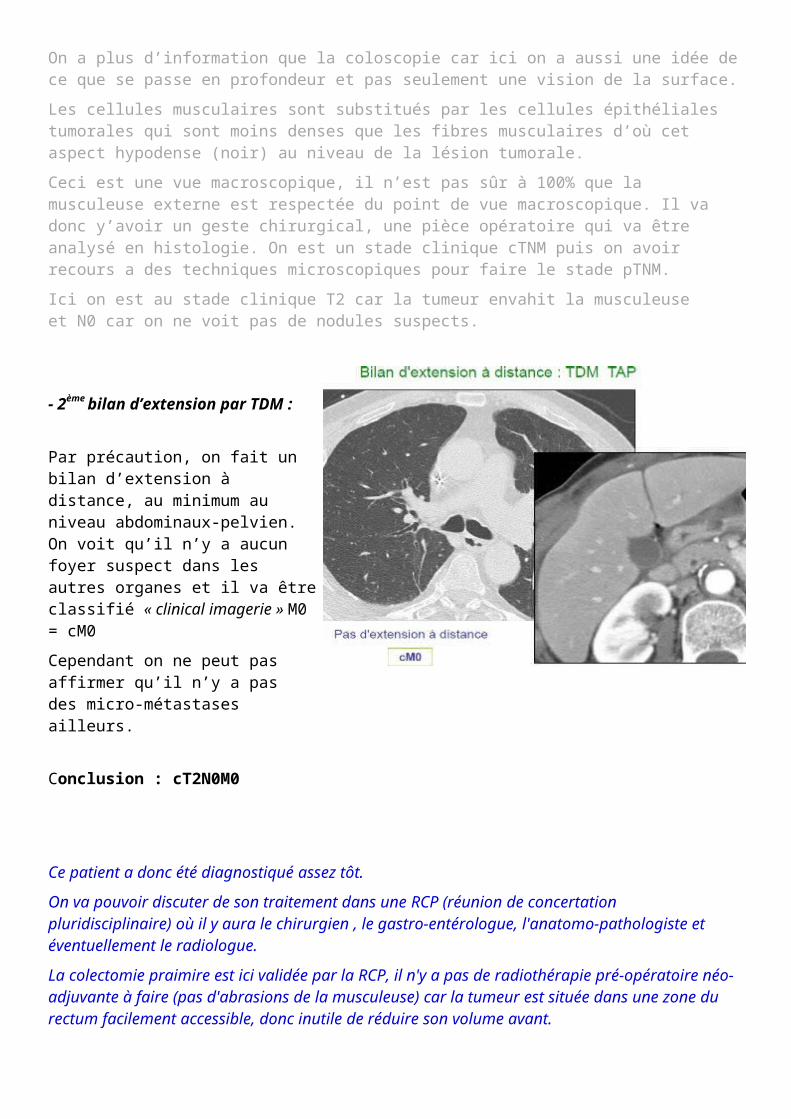
- 2ème bilan d’extension par TDM :
Par précaution, on fait un bilan d’extension à distance, au minimum au niveau abdominaux-pelvien. On voit qu’il n’y a aucun foyer suspect dans les autres organes et il va être classifié « clinical imagerie » M0 = cM0
Cependant on ne peut pas affirmer qu’il n’y a pas des micro-métastases ailleurs.
Conclusion : cT2N0M0
Ce patient a donc été diagnostiqué assez tôt.
On va pouvoir discuter de son traitement dans une RCP (réunion de concertation pluridisciplinaire) où il y aura le chirurgien , le gastro-entérologue, l'anatomo-pathologiste et éventuellement le radiologue.
La colectomie praimire est ici validée par la RCP, il n'y a pas de radiothérapie pré-opératoire néo- adjuvante à faire (pas d'abrasions de la musculeuse) car la tumeur est située dans une zone du rectum facilement accessible, donc inutile de réduire son volume avant.
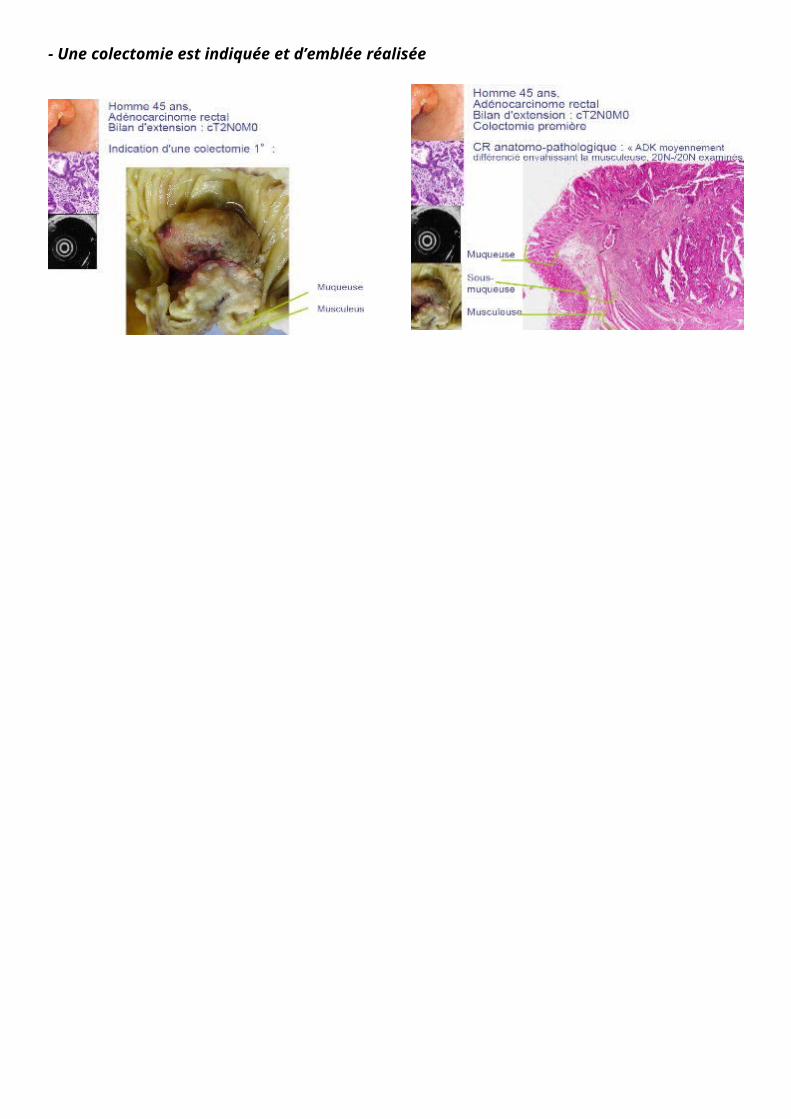
- Une colectomie est indiquée et d’emblée réalisée

La colectomie est réalisée par un chirurgien digestif qui enlève l'ensemble du côlon (qu'on voit ici à l'état frais voir fixé). Il va repérer la tumeur, la mesurer, avec une pièce opératoire qui s'accompagne d'un curage ganglionnaire correspondant aux aires de drainage.
Sur l'histologie fixée avec un simple HES on voit bien la muqueuse normale qui se traduit ensuite en muqueuse dysplasique et une infiltration de la musculaire muqueuse, du plan sous-muqueux, des musculeuses (tunique musculaire lisse du rectum)..
Dans le Curage ganglionnaire 20 ganglions retrouvés négatif, donc pas de métastase ganglionnaire ou à distance.
En anapath, on a une macroscopie à l’état frais, en partie faite par le chirurgien qui va disséquer la lésion. La macroscopie doit être rapide sinon le prélèvement se détériore avant d’être fixé.
À priori ici c’est un état frais, et pas fixé, car on voit la muqueuse, et cette couleur qui ressemble à l’état frais ; et non pas cet aspect jaunâtre quand c’est fixé. On reconnaît la muqueuse et la musculeuse, l’épithélium qui fait des replis, des cryptes, on est dans le bas côlon. C’est donc un épithélium plutôt lisse et un peu cabossé, avec des glandes qui s’invaginent dans la muqueuse.
On reconnaît l’aspect d’un épithélium lieberkhunien, des coupes de glandes +/- transversales, +/- longitudinales. C’est un petit grossissement donc on a du mal à reconnaître. On peut vérifier sur un plus fort grossissement si les cellules tumorales atteignent la musculeuse etc.
Une muqueuse normale : un épithélium de revêtement +/- lisse qui s’invagine de temps en temps, faisant quelques replis, mais pas de grandes valvules conniventes ni toutes ces villosités que l’on voit au niveau de l’intestin grêle. Ici c’est un peu lisse, un peu bosselé et il y a des glandes qui s’invaginent (que l’on voit en coupe longitudinale : doigt de gant/ transversale : aspect de champ de pâquerettes), puis une musculaire muqueuse, (dans le chorion : quelques fibres musculaires lisses, puis il y a la sous-muqueuse et ensuite il y a les 2 couches de fibres musculaires architecture bien organisée vs ici)
confirmation histologique d’une tumeur qui envahit le muscle
Compte-rendu anapath : adénocarcinome moyennement différencié par rapport aux cellules de départ
• Le stade représente le degré d’envahissement en terme tissulaire.• Le grade correspond à la morphologie des cellules, est ce qu’elles ressemblent à l’aspect des cellules de départ.
Il existe maintenant des classifications histo-moléculaires : l’histologie ne suffit plus, il faut des examens moléculaires pour affiner le diagnostic. Les classifications évoluent et varient selon les organes.
On ne nous demande pas de retenir les classifications, juste globalement :
• TA ou TIS : tumeur in situ = intra épithéliale, de bon pronostic (à l’exception de la tumeur de la vessie)
• T1 : envahit la LB et donc le chorion +/- superficiel
• T2 : envahit le muscle
On passe donc de cT2N0M0 a pT2N0 (après anapath)
- Concernant la TNM 8

pTNM est donc plus fin que cTNM
rare que ce soit précisé
On ne fait quasiment jamais de aTNM (très peu d'autopsie au point de vue médical)
Le grade histologie correspond au degré de différenciation, de mitose et d’atypie cellulaire. Plus il est élevé et plus il y a des anomalies.
Parfois le mode de dissémination peut être hématogène, lymphatique (ggl sentinelle etc), mais aussi péri-nerveuse. Les cellules infiltrent la gaine d’un nerf et suivent le trajet du nerf dans l’organisme.
Ce sont des paramètres très peu utilisés. Si un envahissement a lieu, on a va plutôt le rédiger dans le compte-rendu de toute lettres.
En fonction du grade, du stade et du TNM, on fait un super stade combiné. On aura alors différents stades plus globaux.
cancer colo-rectal (TNM7)
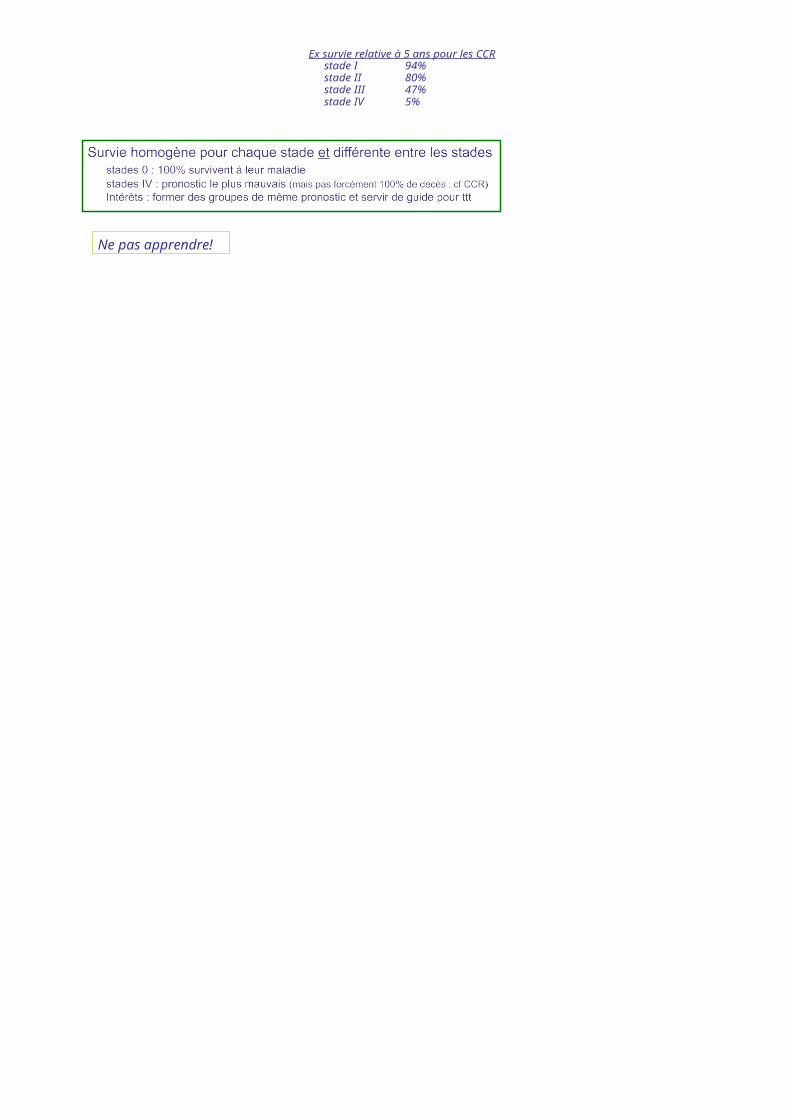
Ex survie relative à 5 ans pour les CCRstade I 94%stade II 80%stade III 47%stade IV 5%
=Stade 1
Ne pas apprendre!

- QCM 3 :
Intérêts principaux de la classification TNM : pour la thérapeutique (donne un stade à la maladie pour savoir si on donne un traitement local, général..) et pour donner un pronostic (stade localisé, généralisé..).
Chaque spécialiste a son propre TNM.
Alors pour la suite Merlio n'a pas du tout détaillé et expliqué comme Capellen donc je vous retranscrit ses explications telles quelles.
Que doit-on faire comme test de génétique somatique chez ce patient ?
Il faut faire une recherche d'instabilité des microsatellites (MSI) car l'Institut national du cancer a décidé de dépister chez les malades qui ont le cancer colo-rectal avant 60 ans si le cancer survenait sur une prédisposition familiale qu'on appelle heriditari non polyposis colo-rectal cancer (HNPCC = Syndrome de Lynch). Ces maladies héréditaires qui prédisposent à l'apparition du cancer colo-rectal touchent moins de 10% des patients qui ont le cancer colo-rectal.
Si on trouve chez un sujet qu'il a une prédisposition génétique on va pouvoir avec l'accord du sujet prévenir la famille et cibler le dépistage précoce du cancer colo-rectal.
Il s'agit donc de dépister ce mécanisme qu'on appelle syndrome d'HNPCC/syndrome de Lynch qui est une prédisposition génétique par mutation des gènes qui réparent/stabilisent l'ADN au niveau des séquences microsatellitaires.
On va rechercher ce phénotype MSI par 2 approches : d'abord IHC puis biologie moléculaire.
L'IHC va nous montrer sur coupe en paraffine la perte d'expression somatique (= dans la tumeur) des protéines de réparation, comme ici par exemple la perte du tandem MLH1 et PMS2 alors que le tandem MSH2 et MSH6 reste positif.
Ce qui est intéressant de noter c'est que les cellules normales gardent les propriétés de réparation de l'ADN alors que les cellules anormales ont perdu ces propriétés. Cela s'explique car dans la famille de ce sujet il y a un élément sur le premier allèle (paternel ou maternel) qui va inactiver le gène et que dans la tumeur est arrivée la perte du 2ème allèle.
Il s'agit d'une transmission d'une mutation inactivatrice d'un des gènes qui code pour une de ces 4 protéines et quand 1 de ces 4 gènes est muté la protéine qui fait l'hétérodimère avec la protéine mutée est aussi disparue.

Donc on a soit une disparition des hétéro-dimères MLH1/PMS2 soit des hétéro-dimères MSH2/MSH6.
On peut donc dire que le phénotype MSI en IHC est lié à l'inactivation de MLH1 ou PMS2.
Si le test IHC peut pas nous dire si c'est une maladie héréditaire, on va confirmer ça par la biologie moléculaire.
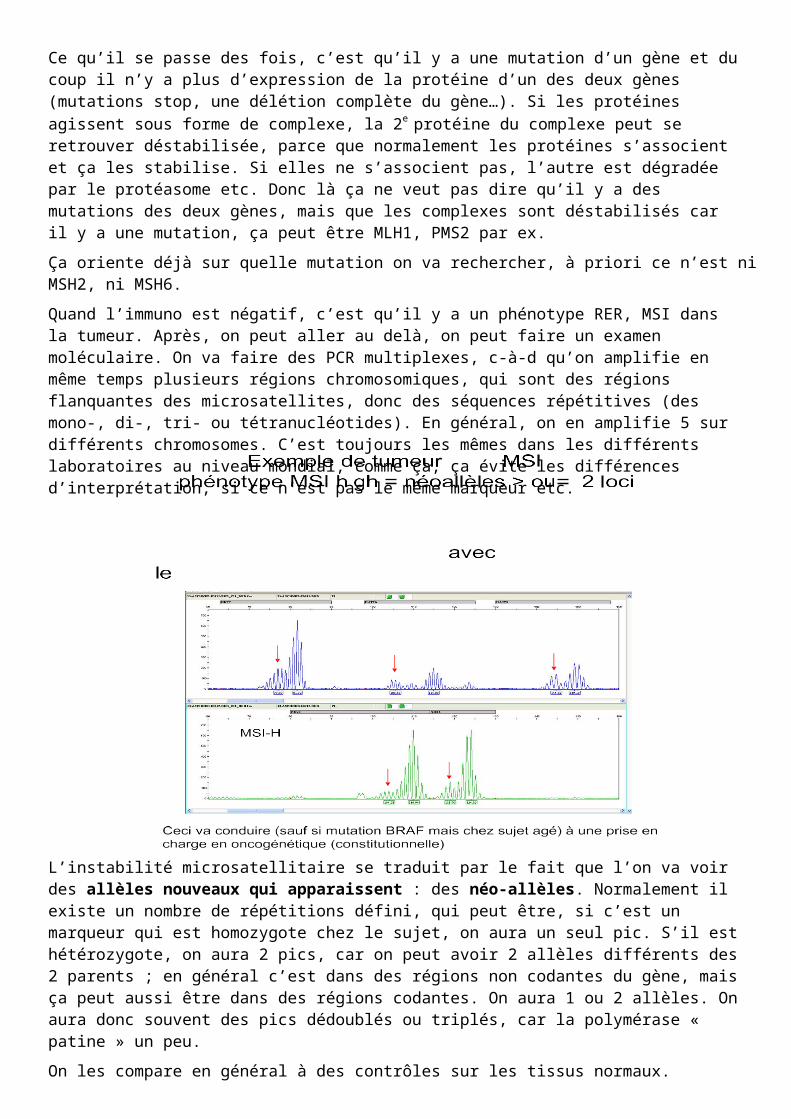
On prend l'ADN tumoral et on regarde s'il y a une instabilité des microsatellites c'est à dire des néo-allèles.
Normalement ces marqueurs sont des marqueurs polymorphes dans la population générale, ils vont donner ces pics en courbe de Gauss. Quand on a une instabilité satellitaire on a des néo-allèles symbolisés par des flèches : là on a 5 marqueurs sur 5 donc on est sûr qu'il y a instabilité.
Mais attention ça ne veut pas forcément dire que c'est héréditaire : épigénétique possible d'où l'importance de la distinction entre maladie constitutionnelle et somatique.
Dans les maladies constitutionnelles an fait il s'agit d'un phénotype avec perte immunohistochimique (statut MSI high) :ces malades ont une mutation germinale des gènes MRR.
Donc l'IHC va permettre en oncogénétique de voir des mutations dans les cellules sanguine et non plus dans la tumeur des gènes MLH1, PMS2...
A côté de la maladie héréditaire dite de Lynch qui prédispose à des cancers chez le sujet jeune on peut avoir dans le cancer colo-rectal des phénotypes MSI sporadique qui sont liés à la méthylation du promoteur du gène MLH1. Dans 10-15% des cancers sporadiques on a le même phénotype (en terme de perte immunohistochimique) avec le statut MSI high mais ce n'est pas une mutation transmise : on ne retrouvera rien au niveau de l'ADN constitutionnel du sujet mais on retrouvera dans la tumeur une méthylation somatique des îlots CPG du promoteur MLH1 ( et dans 40% des cas une mutation BRAF).
Donc avant d'alerter la famille en leur disant qu'il y a une prédisposition familiale il est important de rechercher le phénotype MSI et s'il est positif il faudra aller plus loin avec la biologie moléculaire et la recherche de la méthylation du gène MLH1 et de la mutation du gène BRAF.
Si on se retrouve dans le cas sporadique on ne va pas aller vers l'oncogénétique.
Mais si on ne voit pas une méthylation du gène MLH1 alors qu'on a une perte de MLH1 (phénotype MSI) c'est peut être une maladie de Lynch : consultation en oncogénétique.
Quand on a dépisté la mutation d'un des 4 gènes MRR on rentre dans le cadre de la prédisposition au cancer colo-rectal avec l'intérêt de pouvoir rechercher chez les apparentés s'ils sont porteurs de cette mutation (sans faire de biopsie colorectal, simple prise de sang).
On proposera aux sujets porteurs de la mutation une surveillance par coloscopie tous les ans.
Thérapie pour cancer colo-rectal après chirurgie et chimiothérapie ?
Anticorps bloquant anti-EGFR (important) : Cetuximab
Qu’est-ce qui conditionne le traitement par cet Ac ?
• Il faut que KRAS-NRAS ne soit pas muté (wild-type).
• Seulement chez les stades localement avancés et métastatiques.
Notre patient est au super stade 1 avec 95% de survie.
Dans l’absolu on pourrait lui donner ce traitement, cependant l’AMM régule l’attribution du médicament : localement avancé, métastatique ou wild-type.
Pourquoi ne pas donner ce traitement à ce stade ?
À ce stade on a d’autre arsenal thérapeutique, on utilisera ce traitement seulement si dégradation de l’état.
Si on traite très tôt des lésions peu agressives on peut sélectionner des cellules résistantes (comme pour les antibiotiques) et si le patient progresse et récidive dans sa maladie, on n’aura plus de traitements dispo après.
On a vu qu’on recherche K-RAS.
Pourquoi ne recherche-t-on pas l’expression de l’EGFR ou une mutation de l’EGFR ?
• On ne recherche pas l’expression, car elle ne conditionne pas le traitement. Si l’on veut mettre un médicament qui bloque une molécule, on va voir si cette molécule est exprimée. Du coup, pour qu’un médicament agisse, il faut que sa cible soit présente dans les cellules en question. Au début, on cherchait en IHC si les cellules exprimaient l’EGFR. Puis globalement on remarque que beaucoup de cellules expriment l’EGFR, à peu près toutes, donc ce n’était pas discriminant.
Le mécanisme d’activation dans ces voies colorectales ce n’est pas forcément une surexpression de l’EGFR mais des mécanismes de type plutôt autocrine. Par exemple, la cellule va surexprimer un ligand et du coup aura une boucle autocrine ligand-récepteur. Rechercher l’expression du récepteur n’amène pas d’indications si les cellules dépendent ou pas de cette voie.
• On ne recherche pas de mutation de l’EGFR, car elles sont extrêmement restreintes à certains types de cancers bronchiques. Il n’y a pratiquement pas de mutation de l’EGFR dans d’autres cancers et notamment dans le colorectal, on n’en a jamais trouvé.
• Les recherches de mutation de KRAS : ce n’est pas une tumeur localement avancée ou métastatique, donc on se garde ça pour ultérieurement.
• BRAF : pas forcément non plus, parce que ce n’est pas une tumeur localement avancée ou métastatique.
BRAF quand il est muté, répond à un médicament : le Vémurafénil, prescrit au départ pour les mélanomes, car il y a beaucoup de mutations de BRAF dans les mélanomes. Il y en a aussi dans les cancers colorectaux. Du coup s’il était muté BRAF, il ne serait pas éligible pour le Cetuximab mais pourrait être éligible pour le Vémurafénil. Encore une fois, on se garde ces traitements pour les stades localement avancés ou métastatiques.
Pourquoi recherche-t-on une instabilité microsatellitaire ?
Le patient a 45 ans, cancer chez un patient relativement jeune, alors que les cancers colorectaux sont plus souvent chez les gens plus âgés. On peut donc s’interroger si c’est une tumeur sporadique ou une tumeur héréditaire, qui rentre dans le spectre des syndromes de Lynch, qui sont liés à des mutations de gène de réparation des mésappariements de l’ADN ou mismatch repairs.
Les mutations de ces gènes se traduisent à l’échelle somatique ou à l’échelle héréditaire par une instabilité des séquences répétitives. On appelle cela le phénotype MSI ou MicroSatellite Instability ou RER pour Replication Errors. On va rechercher alors systématiquement chez les sujets jeunes s’il y a ce phénotype RER ou MSI, qui pourrait alors amener vers une consultation de génétique pour voir s’il n’y a pas des mutations germinales dans la famille etc, et que ça ne rentre pas dans le cadre d’un cancer héréditaire. Dans ce cas là, il y aurait une certaine surveillance à apporter, pas uniquement au patient mais à toute la famille, à faire un test de dépistage.
La première technique pour aller voir ces phénotypes RER, MSI est un screening d’IHC. Les mutations touchent des gènes suppresseurs de tumeurs ; les gènes du mismatch repair sont les équivalents des gènes mut HLS du système d’excision des mésappariements chez la bactérie E.Coli. Du coup, on a trouvé des orthologues, c-à-d des gènes homologues chez l’humain : MSH2, MSH6, MLH1, PMS2…
Dans le syndrome de Lynch, c’est une mutation en général d’un de ces gènes. Parfois il y a des choses un peu plus subtiles, des épi mutations qui causent une méthylation aberrante du gène MLH1 etc, mais c’est très rare. Ce sont des mutations de type empreinte génomique aberrante du gène MLH1.
La plupart du temps, ce sont des mutations germinales, c-à-d à l’état hétérozygote chez les patients. Et c’est un gène suppresseur de tumeur donc normalement il faut la perte du 2e allèle, mais malheureusement chez ces patients qui ont une mutation germinale dans toutes les cellules de l’organisme, (quelque soit le gène concerné) à un moment donné, il y aura une cellule qui va perdre le 2e allèle et qui va du coup se retrouver nullizygote pour la fonction du gène. On aura à ce moment une perte de fonction et ces cellules vont accumuler un certain nombre de problèmes. Soit c’est une mutation de gènes impliqués dans le cycle cellulaire, comme rétinoblastome, p53 (gate-keeper : barrière du cycle cellulaire) etc ; et la cellule va commencer à cycler n’importe comment.
Ici ce sont des réparations de l’ADN qui vont être déficientes et du coup les cellules vont accumuler des mutations. Donc ces gènes on les appelle des care-taker : ils ont un rôle de suppresseur de tumeur mais indirect, car leur mutation ne crée pas directement une prolifération cellulaire anormale ; mais leur mutation va permettre, au hasard, la survenue d’autres mutations. (les réparations d’ADN ne se faisant pas correctement ; au hasard, la réplication va mal se faire, il y aura une mutation qui va ne pas être détectée et va se perpétuer au lieu d’être réparée. À ce moment là, ça va toucher d’autres oncogènes, les gènes suppresseurs de tumeur qui eux ont un impact direct sur le cycle cellulaire etc).
Comment teste-t-on cela ?
Comme ce sont des mutations de perte de fonction, on va rechercher la perte d’expression des protéines, tout simplement, en première intention. On fait un screening en IHC. L’IHC, comparée à la FISH, n’a pas de contrôle interne. C-à-d que la FISH, soit on a une hybridation normale, soit on a une hybridation anormale, soit on n’a pas d’hybridation. Si on n’a pas d’hybridation, on ne peut pas interpréter, et si on a une hybridation anormale, on va voir le type d’anomalie.
En IHC, on n’a pas cette chance, car si on veut chercher une surexpression ou une sous expression mais qu’on n’a pas un contrôle positif sur la lame du même tissu. On peut avoir un faux positif si c’est du bruit de fond, ou un faux négatif, s’il y a une surexpression mais on ne la voit pas, ou bien s’il y a une expression et qu’on ne la voit pas parce que la lame est mal fixée, donc on ne voit pas d’expression car l’Ag est dégradé.
Là on a de la chance, car sur les glandes normales, à proximité de la tumeur, ces protéines sont positives. On a un contrôle interne. Du coup, on voit très bien sur les glandes normales l’expression de MLH1 en immuno peroxydase de PMS2 etc. Par contre dans la tumeur, on ne voit pas leur expression, mais on voit quand même MSH2 et MSH6. Parfois on a des pertes combinées parce que les protéines agissent sous forme de complexe. Cela ne veut pas forcément dire qu’il y a une mutation des deux gènes.

Ce qu’il se passe des fois, c’est qu’il y a une mutation d’un gène et du coup il n’y a plus d’expression de la protéine d’un des deux gènes (mutations stop, une délétion complète du gène…). Si les protéines agissent sous forme de complexe, la 2e protéine du complexe peut se retrouver déstabilisée, parce que normalement les protéines s’associent et ça les stabilise. Si elles ne s’associent pas, l’autre est dégradée par le protéasome etc. Donc là ça ne veut pas dire qu’il y a des mutations des deux gènes, mais que les complexes sont déstabilisés car il y a une mutation, ça peut être MLH1, PMS2 par ex.
Ça oriente déjà sur quelle mutation on va rechercher, à priori ce n’est ni MSH2, ni MSH6.
Quand l’immuno est négatif, c’est qu’il y a un phénotype RER, MSI dans la tumeur. Après, on peut aller au delà, on peut faire un examen moléculaire. On va faire des PCR multiplexes, c-à-d qu’on amplifie en même temps plusieurs régions chromosomiques, qui sont des régions flanquantes des microsatellites, donc des séquences répétitives (des mono-, di-, tri- ou tétranucléotides). En général, on en amplifie 5 sur différents chromosomes. C’est toujours les mêmes dans les différents laboratoires au niveau mondial, comme ça, ça évite les différences d’interprétation, si ce n’est pas le même marqueur etc.
L’instabilité microsatellitaire se traduit par le fait que l’on va voir des allèles nouveaux qui apparaissent : des néo-allèles. Normalement il existe un nombre de répétitions défini, qui peut être, si c’est un marqueur qui est homozygote chez le sujet, on aura un seul pic. S’il est hétérozygote, on aura 2 pics, car on peut avoir 2 allèles différents des 2 parents ; en général c’est dans des régions non codantes du gène, mais ça peut aussi être dans des régions codantes. On aura 1 ou 2 allèles. On aura donc souvent des pics dédoublés ou triplés, car la polymérase « patine » un peu.
On les compare en général à des contrôles sur les tissus normaux. L’idéal est d’avoir l’ADN normal du même patient, mais ce n’est pas obligatoire. Si on voit des petits pics à droite à gauche, cela correspond à des microinsertions ou des microdélétions, c-à-d des répétitions en moins ou en plus de la séquence répétitive.
On va alors scorer le MSI (Micro Satellite Instablity) en low, medium, high. Si le patient a des instabilités sur tous les marqueurs testés, il va être classé MSI high. Dans ces cas là, il y a une chance que ce soit un cancer héréditaire. Cela signifie que l’instabilité microsatellitaire était là dès le début de l’oncogenèse, c’est le mécanisme qui l’a déclenché. Il avait une mutation germinale, une perte de l’allèle wild type, et les tous premiers événements oncogéniques ont été l’instabilité microsatellitaire, l’instabilité des séquences de réplication. Cette dernière se traduit par l’instabilité des séquences répétitives mais aussi par l’apparition de mutations dans les séquences codantes des gènes ; parce que si on ne mute que les microsatellites qui sont dans des régions non codantes, il n’y aura pas de phénotype tumoral.
Par ailleurs, on a d’autres facteurs pour savoir si c’est un cancer héréditaire.

La mutation BRAF n’est retrouvée que dans les cancers sporadiques, c-à-d pas dans les cancers à prédisposition génétique, mais dans les cancers qui apparaissent comme ça, au hasard. Si on retrouve une mutation BRAF, c’est donc un critère d’exclusion d’un cancer héréditaire. À ce moment là, ça peut être MSI high, car la mutation est apparue quand même tôt dans l’oncogenèse.
Question élève : Qu’est-ce qui fait dire que c’est un cancer héréditaire ?
Ce qui fait dire que c’est un cancer héréditaire, c’est que tu as tous les loci que tu as testé qui ont des instabilités. Ce qui veut dire que probablement, cette instabilité s’est mise en place très tôt dans la maladie. Cela laisse donc penser que ça peut être un cancer héréditaire. Si la mutation des gènes de réparation de l’ADN est survenue au cours de la progression tumorale, elle va être survenue plus tard, elle concernera certainement moins de cellules, moins d’allèles… Du coup, on n’aura peut-être pas autant d’allèles mutés. C’est par rapport au nombre de régions chromosomiques qui présentent ces régions d’instabilité.
Conclusion :
Encore une fois, ce n’est pas pour autant sûr que c’est un cancer héréditaire, il y a encore des chances que ce soit un cancer sporadique, mais que la mutation soit survenue assez tôt. BRAF est un critère d’exclusion : si BRAF est muté, c’est un cancer sporadique. Il n’y a pas d’exception trouvée pour l’instant. Il n’y a en fait jamais de mutation de BRAF dans le syndrome de Lynch ; les mutations initiatrices vont toucher d’autres gènes, par ex le récepteur au TGF β, parce qu’il y a une séquence répétitive codante dans le gène du TGF β récepteur 2 qui va être muté en conséquence des mutations du mismatch repair.
Il peut quand même y avoir un certain nombre de cancers sporadiques qui sont MSI+ et qui vont avoir des méthylations au niveau du gène MLH1, c-à-d un mécanisme d’épigénétique d’inactivation qu’on peut rechercher, ainsi que des mutations du gène BRAF.
Il n’existe quasiment jamais de mutation dans le cas d’un cancer colorectal.
Les phénomènes de carcinogénèse ne sont pas les mêmes dans le poumon et le colon donc ce ne sont pas les mêmes gènes qui ont touchés mais parfois certains sont retrouvés dans les deux.
Si la tumeur a évolué, ça peut permettre d’inclure dans des protocoles thérapeutiques le médicament Vémurafénil, qui est un inhibiteur de l’activité sérine-thréonine kinase de BRAF, et qui marche bien sur cette mutation. Elle est utilisée en 1ère intention pour les mélanomes. Il existe un programme : Accès-cible ou Accès moléculaire, qui fait que maintenant on ne traite plus forcément un cancer selon le type histologique tumoral, comme on le faisait auparavant ; parce qu’on avait associé tel type histologique à telle réponse, mais c’était empirique. Maintenant, on a des critères moléculaires, qui nous permettent de dire par exemple : la présence de la mutation V600E signe une sensibilité au Vémurafénil, donc on va traiter. On s’en fout si le patient a un cancer du sein, de la thyroïde, du poumon. S’il est BRAF muté, il peut être traité avec le Vémurafénil et il aura donc éventuellement une bonne réponse s’il n’a pas de mécanismes de résistance qu’il développe de manière concomittante.

L’expression de l’EFGR n’est pas prédictive de la réponse au cetuximab il peut y avoir des sur-expressions mais le plus souvent se sont des sur-expressions de ligands qui vont être impliquées dans le cancer colorectal et puis éventuellement des mutations en aval.
- Point important : Quand on donne une thérapie ciblée, quelle mutation va être décisive pour donner une thérapie ciblée ?
On va vous dire une voie métabolique, un cancer dont on vous a jamais parlé et puis on va vous dire qu’on a une mutation là et un médicament qui agit à tel ou tel niveau, et est ce que ça va marcher ou pas ? Vous pouvez même si vous ne connaissez absolument pas cette voie ou cette pathologie, de manière logique prédire si le traitement va marcher ou pas. Comment ? En fait, la tactique c’est que le médicament doit agir sur la cible ou en aval de la cible. Pourquoi ? C’est un peu logique, si vous avez une cible mutée à ce stade là et que le médicament agit sur la cible mutée et on sait qu’il marche parce qu’il y a une mutation de résistance mais qu’on sait que c’est une mutation qui est sensible au médicament comme je disais pour BRAF. A ce moment là, si on tape sur BRAF, ça va marcher, parce que la cible est mutée et sensible.
Maintenant si on a une molécule comme le cetuximab (Ac), qui bloquent l’EGFR mais qu’on a une voie en aval qui est constitutivement activée parce qu’il y a une mutation de KiRAS ou BRAF, ça sert à rien. C’est un peu comme si vous faisiez le relais 4 fois 400 mètres, le principe c’est qu’on attend que le copain nous refile le bâton pour commencer à courir. Donc si on stoppe le premier coureur, qui du coup ne passe pas le témoin au deuxième, la course s’arrête. Donc si la cible du médicament est le premier coureur, ça fonctionne. Si le deuxième coureur, s’en fou des règles du jeux et qu’il court quand même, même si on lui a pas passé le bâton, et qu’on arrête le premier, ça va pas marcher. Du coup il faut « taper » sur le coureur qui présente la mutation ou bien après. Donc si il y a une mutation, on tape la mutation ou bien après.
Donc en cancérologie, on agit sur la cible qui présente la mutation, si cette mutation est sensible ou en dessous. En dessous, ça peut être problématique car des fois les voies se ramifient et du coup ça va être des fois moins efficace car vous allez être bloqués. Par exemple le Rc EGF est muté (mutation de résistance), du coup on bloque la voie Ras/Raf MAP-kinase, le problème c’est que EGFR ne signalise pas que vers la voie de MAP kinase, il peut aussi signaliser la voie de PI3kinase. Du coup on va bloquer qu’une des voies, donc se sera moins efficace, ou il faudra deux médicaments. On tape toujours au niveau de la mutation si celle-ci
est sensible ou bien après sinon ça ne fonctionne pas. Comme on vient de l’expliquer pour les cetuximab, si on bloque le Rc avec un Ac et si on a la voie d’EGFR et de Ras-MAP kinase et PI3-kinase d’un côté, ça ne marchera pas car la voie est déjà activée en dessous. Le principe est que le Rc fixe son ligand, se dimérise, s’autophosphoryle et recrute les protéines de type Graptou,etc au niveau des tyrosines phosphorylées et puis
il va recruter Ras, l’activer et ça déclenchera la voie des MAP kinase. Donc si il y a une mutation de Ras, Raf, etc en aval, on ne peut pas bloquer en amont. Il faut bloquer au niveau de la cible ou en dessous de la cible mutée.
Ce Rc EGF est impliqué dans plein de maladies, mais pas de la même façon. Par exemple dans les gliomes, c’est des amplifications géniques, dans le colon c’est plutôt des boucles autocrines, dans le cancer du poumon c’est des mutations activatrices du domaine tyrosine kinase, etc. Ce n’est pas les mêmes mécanismes selon les cancers et du coup selon les cancers, les mêmes médicaments ne fonctionneront pas. Par exemple, un inhibiteur de tyrosine kinase ne marche que sur certaines mutations et pas sur les Rc de Wildtype, donc ça ne sert à rien de donner des inhibiteurs de tyrosines kinases sur les tumeurs pour lesquels on sait que le mécanisme d’action ne passe pas par des mutations. C’est pour cela que l’on fait des tests moléculaires.
III. Carcinome mammaire.
Intérêt du dossier : Rappeler et illustrer les facteurs pronostiques et les facteurs prédictifs de la réponse thérapeutique (thérapeutiques ciblées), obtenus à partir de l’analyse bio-pathologique des tissus tumoraux.
Dans le cas des cancers mammaires, il y aussi des mutations génétiques qui peuvent prédisposer à 10% des cancers mammaires, les fameuses mutations des gènes BRC1et BRAC2 (n’en parle pas aujourd’hui).Pour les cancers mammaires, il y a aussi l’interrogatoire « est ce que il y a eu plusieurs cancers mammaires dans la famille ? » (on voit si la mère, la tante la grand mère… l’ont) et en fonction de ça on peut aussi, comme pour le cancer colorectal aller chercher les 10% de cancers mammaires qui sont héréditaires.On a l’exemple d’Angelina joli qui a fait une exérèse des deux seins et des deux ovaires en prévention.
Rappels : le sein normal
Les messiers n’ont que des canaux galactoforiques rudimentaires et les dames ont à la fois des acinis mammaires dans le tissus palléal et du tissu adipeux qui va donner le volume des seins (seins=caractères sexuels secondaires qui va permettre de faire l’allaitement à la naissance).
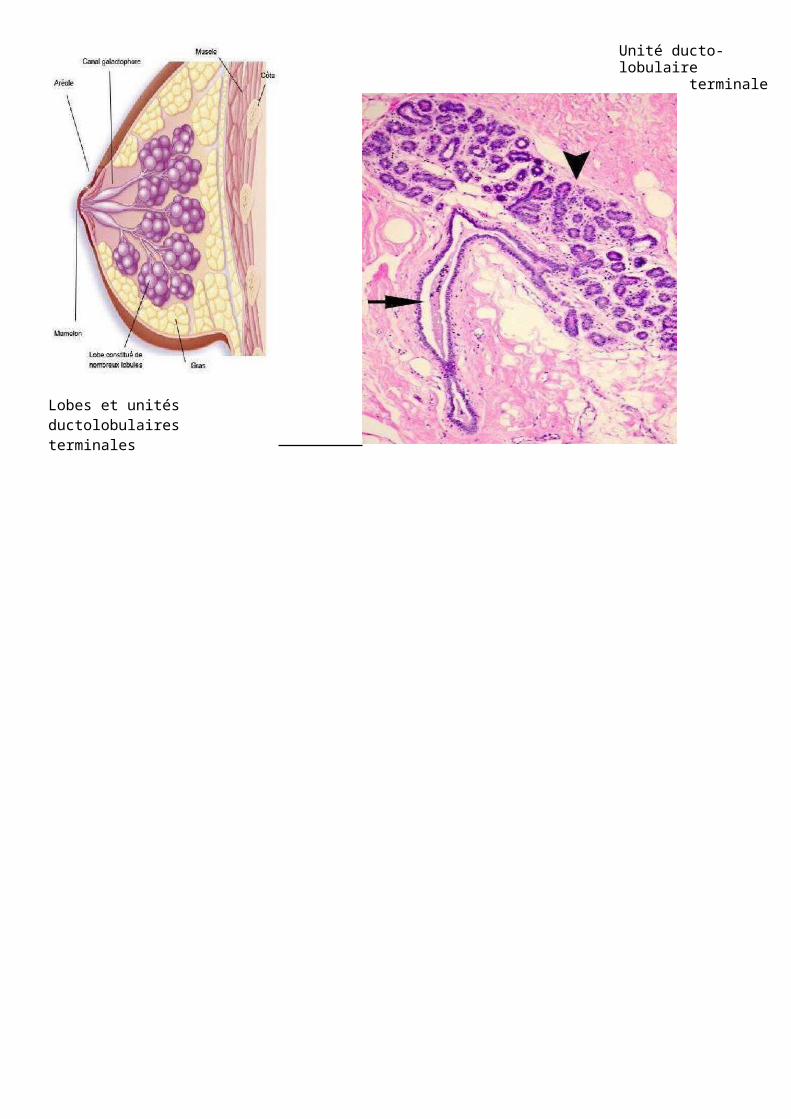
Lobes et unités ductolobulaires terminales
Unité ducto-lobulaireterminale

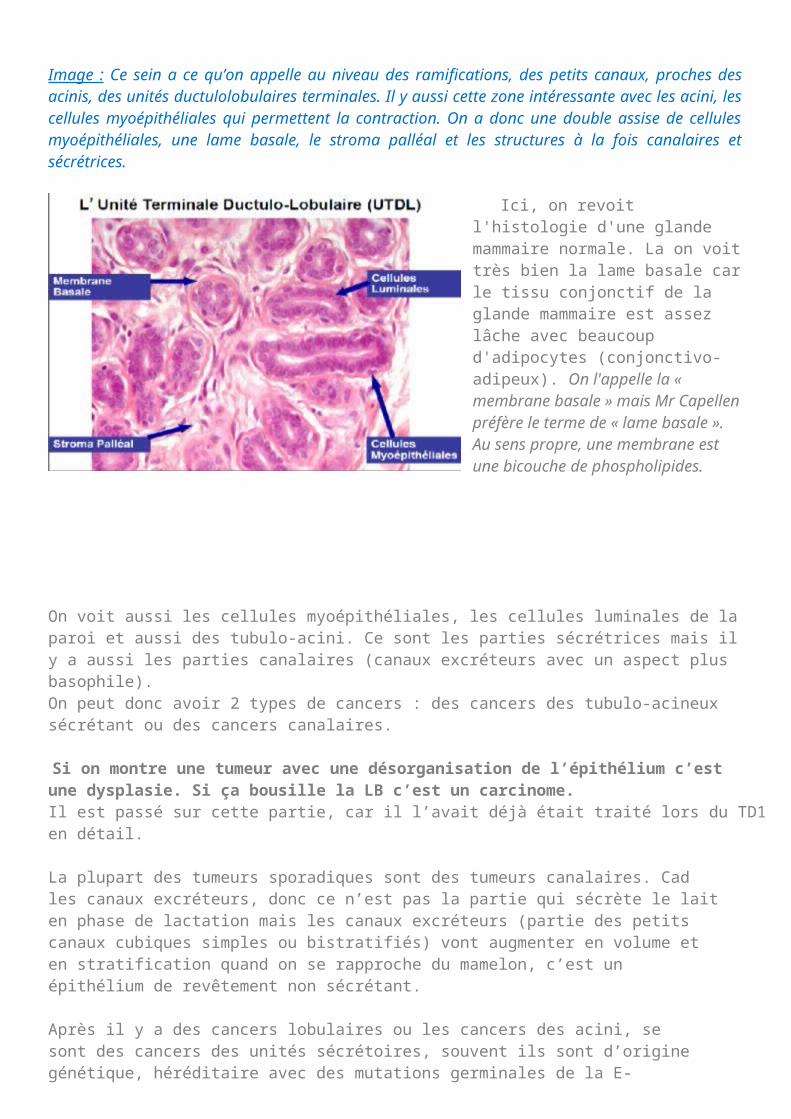
Image : Ce sein a ce qu’on appelle au niveau des ramifications, des petits canaux, proches des acinis, des unités ductulolobulaires terminales. Il y aussi cette zone intéressante avec les acini, les cellules myoépithéliales qui permettent la contraction. On a donc une double assise de cellules myoépithéliales, une lame basale, le stroma palléal et les structures à la fois canalaires et sécrétrices.
Ici, on revoit l'histologie d'une glande mammaire normale. La on voit très bien la lame basale car le tissu conjonctif de la glande mammaire est assez lâche avec beaucoup d'adipocytes (conjonctivo- adipeux). On l'appelle la « membrane basale » mais Mr Capellen préfère le terme de « lame basale ». Au sens propre, une membrane est une bicouche de phospholipides.
On voit aussi les cellules myoépithéliales, les cellules luminales de la paroi et aussi des tubulo-acini. Ce sont les parties sécrétrices mais il y a aussi les parties canalaires (canaux excréteurs avec un aspect plus basophile).On peut donc avoir 2 types de cancers : des cancers des tubulo-acineux sécrétant ou des cancers canalaires.
Si on montre une tumeur avec une désorganisation de l’épithélium c’est une dysplasie. Si ça bousille la LB c’est un carcinome.Il est passé sur cette partie, car il l’avait déjà était traité lors du TD1 en détail.
La plupart des tumeurs sporadiques sont des tumeurs canalaires. Cad les canaux excréteurs, donc ce n’est pas la partie qui sécrète le lait en phase de lactation mais les canaux excréteurs (partie des petits canaux cubiques simples ou bistratifiés) vont augmenter en volume et en stratification quand on se rapproche du mamelon, c’est un épithélium de revêtement non sécrétant.
Après il y a des cancers lobulaires ou les cancers des acini, se sont des cancers des unités sécrétoires, souvent ils sont d’origine génétique, héréditaire avec des mutations germinales de la E-cadhérines notamment du gène de la E-cadhérine qui est une molécule épithéliale d’adhésion cellule-cellule.
Comment une molécule d’adhésion peut-être un gène d’oncogénèse ?En fait, ça peut-être un gène qui quand il est perdu va aboutir à un changement morphologique des cellules, une transition épithéliomésenchymateuse etc. Ca va également faciliter la transformation tumorale puisque ces cellules vont perdre leur capacité adhésives, etc quand il y a une mutation qui est hétérozygote au départ mais il y a aura forcément le deuxième allèle qui va sauter dans les cellules tumorales dans certaines cellules somatiques. Donc à ce moment là si certaines cellules subissent après d’autres mutations etc, elles vont avoir plus de capacités à se transformer en cellules tumorales parce qu’elles ont déjà fait la transition épithélio- mésenchymateuse.

Voici le carcinome infiltrant qui infiltre le tissu paléaire. On le verra très bien sur une mammographie, examen de dépistage. On a aussi des cancers lobulaires in situ qu’on peut repérer également sur la mammographie, et notamment le canalaire in situ, qui s’accompagne assez souvent de microcalcifications.
Historique rapide du sein :On a une dissémination lymphatique, malheureusement on a souvent une atteinte des ganglions axillaires, du coup c’est un facteur de mauvais pronostic. On peut avoir aussi des métastases infracliniques, cad qui ne sont pas détectables au départ dès le diagnostic, elles sont présentes dans 25% des cas. Donc elles sont là mais on ne les voit pas et elles vont se réveiller à un moment donné et donner une aggravation du pronostic dans la maladie.On peut guérir d’un cancer métastatique mais c’est une médiane. Sur l’ensemble de la population de patientes, la médiane c’est entre 2 et 10 ans le décès pour les patientes qui ont les métastases les plus difficiles sur le plan thérapeutique. C'est une moyenne, certaines patientes vont survivre très longtemps et vont décéder d'une autre cause pas forcément liée à leur cancer comme le vieillissement. Elles seront quand même porteuses de cellules tumorales résiduelles mais ne mourront pas du cancer à proprement dit.
Ca va être variable en fonction de l’évolution des métastases (est ce qu'elles sont autonomes ? Comment elles prolifèrent ?etc...), des caractéristiques biologiques de celles ci, si elles répondent ou pas au traitement et de leur extirpabilité (sont-elles ré-sécables chirurgicalement si ce sont des métastases avec des localisations embarrassantes comme le cerveau ou le foie).
Question élève 2015-2016 : Qu'est ce que la maladie de Paget ?Réponse Prof : C'est une maladie chronique au niveau du sein avec des métaplasies et qui peut parfois aboutir à un cancer.
Concernant la distinction entre canalaire et in situ : le terme in situ signifie que le carcinome reste dans l'épithélium, on a une hypertrophie de l'épithélium mais la tumeur reste intra-épithéliale (elle n'envahit pas la lame basale). A partir du moment où la tumeur envahit le stroma, on parle de carcinome infiltrant.

1. Histoire naturelle du carcinome infiltrant du sein.
L’histoire naturelle du cancer infiltrant du sein, c’est que c’est une tumeur primitive qui vient du canal du lobule, qui va pouvoir, lorsqu’elle est infiltrante, disséminer par voie lymphatique vers le fameux ganglion sentinelle, et par voie sanguine avec une atteinte des ganglions du creux axillaire dans 40% des patientes opérées, c’est extrêmement fréquent.Au moment de la chirurgie initiale, on estime qu’il y a 25% des cas qui ont des métastases non décelées. C'est-à-dire que le bilan est normal et puis qu’on va révéler assez rapidement le fait qu’il y ait des métastases. Les métastases sont localisées au niveau des os, du foie, des poumons, du cerveau, etc.Le temps moyen entre la découverte de la métastase et le décès va de 2 à 10 ans et va être très variables en fonction de l’évolutivité des métastases, avec le fait qu’on puisse les enlever et qu’elles soient ou non répondeuses au traitement systémique. Donc globalement 40% des gens qui ont des métastases ganglionnaires, peut être même 25% de plus de gens qui n’ont pas de métastases visibles mais qui en aurait (c’est assez fréquent) mettent un espoir dans la réponse de ces métastases aux traitements systémiques.
→ Tumeur primitive au niveau du sein.→ Dissémination par voies lymphatique ou sanguine.→ Atteinte des ganglions du creux axillaires dans 40 % des cas.→ Au moment de la chirurgie initiale, on aura présence de métastases non décelables cliniquement et par imagerie à distance de la tumeur dans 25 % des cas.En fait on ne voit rien, mais il y a quand même des métastases « occultes ». C'est donc une tumeur qui peut être assez agressive dans ces cas là.→ Les sites métastatiques : os, foie, poumons, cerveau, peau, etc...
2. Les facteurs pronostiques et les facteurs prédictifs dans le cancer du sein.
Facteurs pronostics :→ Risque de rechutes locales, métastatiques.→ Risque de décès.
Facteurs prédictifs : capacité de réponse tumorale à un traitement médical d'induction (chimio, hormono, bio) : NEOADJUVANT Influence du traitement médical ADJUVANT sur la survie des patientes opérées d'emblée

Influence du traitement médical PALLIATIF sur la survie des patientes métastatiques
Quels sont les facteurs pronostics ou prédictifs dans le cancer du sein :
Les facteurs pronostics : donnent le risque de rechute locale ou le risque de métastase ou le risque de décèsLes facteurs prédictifs : évaluent la réponse à un traitement
Soit à une chimiothérapie néoadjuvante ou adjuvante soit à un traitement ciblé
Facteurs pronostics :Les facteurs pronostics sont les risques de rechute locale et métastatique. Quand on voit que ça rechute localement et que ça métastase, c’est un facteur de mauvais pronostic et donc ça va augmenter le risque de décès.-> Un facteur pronostic est un facteur qui va donner une indication sur la gravité de maladie dans l’absolu. On va dire le stade, le grade, la présence de métastases, etc. Tout cela se sont des facteurs pronostics parce que l’on sait que la tumeur va être globalement plus grave quand il y a présence de ces caractéristiques là. Un facteur pronostique va donner une idée de l'évolution de la maladie quelque soit le traitement appliqué, ça peut être un facteur pronostique histologique, de biologie moléculaire, etc...→ Risque de rechutes locales, métastatiques.→ Risque de décès.Ces facteurs vont fournir un renseignement sur « l’après » chirurgie.
Facteurs prédictifs :-> Un facteur prédictif est un facteur qui peut être pronostic aussi. Parfois, c’est inverse, des fois la valeur pronostic et prédictive n’est pas dans le même sens. Un facteur prédictif c’est quand on va avoir une évolution particulière de la tumeur mais dans un contexte d’un traitement bien particulier. C’est la capacité de réponses à un traitement particulier qui peut être une chimiothérapie, une thérapie ciblée, une hormono thérapie, etc. Donc la présence de ce biomarqueur va nous dire si par exemple il faut que les cellules soient positives pour les Rc pour répondre aux inhibiteurs hormonaux. Il faut qu’il y ait la présence de l’expression du Rc Rb2 pour répondre aux thérapies avec des Ac bloquants un peu par analogie avec le cetuximab pour l’EGFR et il y a la herceptin pour Rb2 dans le cancer du sein.Pour la chimiothérapie, généralement, le facteur plus ou moins prédictif est l’index mitotique des cellules, cad le marquage au ki-67 en IHC.
2. Types histologiques.
- Canalaires 80 à 85 % (structures excrétrices sans propriétés sécrétoires)

- Lobulaires 10 à 15 %- Autres 5 %A partir de l’histologie normale du sein, on va faire la classification des cancers du sein. Ces cancers peuvent être :
soit des cancers des petits canaux : 80% des cas soit des cancers des acini : globalement 15% des cas les autres : 5% des cas
On va classer les cancers selon le canal ou selon l’acinus. On parlera donc de cancer canalaire ou lobulaire. Ensuite on va classer ce cancer en fonction de son caractère invasif ou non, est ce qu’il a franchi la lame basale ou est ce qu’il est in situ. On aura donc des carcinomes lobulaires in situ ou des cancers canalaires in situ CCIS ou des cancers canalaires infiltrant et des cancers lobulaires infiltrant
Si on prend une cellule acineuse et canalaire sur une morphologie normale, elles n’ont pas les mêmes types de colorations sur le plan histologique. Les canalaires sont davantage basophiles, se sont les cellules sécrétoires du coup elles vont sécréter un certain nombre de sécrétion lipidique et du coup ça prend pas les colorants.Tout cela pour montrer que les différences moléculaires dans ces cancers souvent dues à la perte d’expression de la E-cadhérine. La plupart des mutations sont des mutations stop et du coup se sont des mutations qui tronquent la protéine à un certain niveau. Soit il manque un bout de la protéine, soit la protéine n’est pas du tout exprimée car en général quand il y a un codon STOP dans la protéine, elle est instable, elle n’est même pas présente à la surface des cellules, on a pas la queue intracytoplasmique, on a rien du tout. La partie extracellulaire sont des protéines de la famille des CAM domaines à Ig Calcium dépendante et donc elles se fixent par interaction homophile au niveau extracellulaire (cellule-cellule). Dans les cancers lobulaires, souvent, soit de manière sporadique, soit de manière germinale, on a une perte de fonction, d’expression par différents mécanismes. Ces derniers peuvent être de manière épigénétique par méthylation des promoteurs, des mutations avec des codons STOP (c’est le plus efficace pour inactiver les protéines). Les mutations surviennent au hasard dans les cellules, mais celles qu’on trouve au niveau clonal dans la tumeur, c’est celles qui ont donné un avantage particulier à la cellule tumorale et du coup c’est une sélection presque Darwinienne des mutations les plus efficaces pour le processus en question. Donc dans les cancers, les codons STOP sont les plus efficaces car ils bousillent la protéine du gène suppresseur de tumeur.
Dans les cancers canalaires ce ne sont pas les mêmes mécanismes moléculaires, du coup généralement, on n’a pas la perte d’expression de la E-cadhérine qu’on peut voir en IHC également.
Pour revenir sur la biologie de ces cancers, on va opposer les carcinomes canalaires infiltrants, où sont respectés les systèmes de jonction, que l’on appelle les Ecadhérines, aux cancers lobulaires in situ, où sont perdu les Ecadhérines. Pour reconnaitre ces deux types de cancers on va faire notamment une immunohistochimie avec les Ecadhérines.

A Gauche, on a un cancer canalaire infiltrant. On voit que l'épithélium prolifère n'importe comment, il est totalement désorganisé. On a peut avoir des canaux normaux. Ce marquage est intéressant car on a un contrôle positif sur la lame ce qui n’est pas toujours le cas. C’est un peu comme les protéines du mismatch repair, où les cellules normales présentes sur la tumeur épithéliale vont toujours être positives. Donc si on a une absence de marquage et qu’on a des cellules positives à côté, on sait que ce n’est pas un problème pré- analytique, une mauvaise fixation, une mauvaise technique IHC ou histologique, c’est que vraiment la tumeur est là. Les cellules sont basophiles donc structures canalaires. Souvent ces tumeurs sont E-cadhérine positives, en revanche les cancers lobulaires infiltrants sont caractérisés par des cellules plus éosinophiles.
A droite on a un canal normal marqué à l'E-cadhérine ; dans le cas des cellules tumorales, elles sont E- cadhérine négatives. Les cancers lobulaires sont donc généralement E-cadhérine négatifs, ce qui démontre souvent une perte d'expression des E-cadhérines dans ce type de cancers.
4. Facteurs pronostiques dans le cancer du sein.
Facteurs pronostiques cliniques : permettent de définir l’agressivité du cancer, le risque de métastases, le risque de décès en fonction de plusieurs paramètres qui sont :
→ L'âge, plus le cancer surviendra jeune plus il sera agressifSi le cancer apparaît tôt dans la vie c'est plus souvent de mauvais pronostique.)
→ La grossesse, faut pas oublier de palper les seins chez les femmes enceintes pendant la grossesse. Souvent la grossesse est un facteur d’aggravation du cancer du sein qui flambe parce que la femme est enceinte du fait de l’imprégnation hormonaleUne femme qui a eu des grossesses simples ou multiples aura un pronostique plutôt favorable. On pense que c'est dû à l'imprégnation hormonale, mais on ne sait pas très bien pourquoi.
→ Le stade cTNM : les facteurs pronostics classiques qui sont le CTNM Le stade cTNM, plus le stade est avancé plus le pronostique sera mauvais.
Facteurs pronostiques histologiques : vont permettre de donner un grade au cancer du sein. C’est tout ce qui est grade histologique, type histologique et puis la présence d’embole.
→ Atteinte des ganglions axillaires.→ La taille tumorale histologique / l’atteinte des ganglions→ Le grade histologique

→ Type histologique→ Emboles vasculaires péritumoraux
le stade histologique. Plus les cellules sont indifférenciées, plus c’est de mauvais diagnostics.→ Le type histologique, certains types histologiques sont de meilleurs pronostiques (cancer à cellules différenciées qui sécrètent leur contenu habituelle, biologie proche des cellules normales) alors qu'à l'opposé un cancer à composante inflammatoire sera de plus mauvais pronostiques. Il y a dans ce cas là production de cytokines par les cellules inflammatoires et réaction entre le stroma et l'épithélium.→ Présence d'emboles vasculaires péri tumoraux, en général de mauvais pronostique car c’est un signe de propagation hématogène probable avec des métastases à distance. En général, à partir d'un stade, la tumeur a besoin de nouveaux vaisseaux pour croître sous peine de rentrer en hypoxie et en nécrose. Statistiquement, s’il y a présence d'emboles vasculaires péri tumoraux, il y a de grandes chances pour qu'il y ait une dissémination hématogène.
Ganglions axillaires (N) Ne pas s'attarder sur les pourcentages de survie !
Ex pour nous montrer quelques éléments pronostics : Quand on a des ganglions, si on a par ex PN0 : la survie globale à 10ans est de 75%.Si on a un ganglion atteint, elle passe à 25%, donc facteurs pronostic majeur.
La taille d’une tumorale, si on a une petite tumeur, d’où l’intérêt de la mammographie, à 5ans 91% des malades sont en vie.Si on a une grosse tumeur qu’on a négligé d’aller faire une mammographie de dépistage, 60% de survie à 5ans (donc 40% de malade qui décèdent pendant les 5premières années).
Survie globale à 10 ans :→ pN0 (aucuns ganglions positifs) environ 75 % de survie.→ pN1 + (au moins un ganglions positif) 25 à 30 % de survie.

Un ganglion a l’aspect d’un rein, on a les vaisseaux lymphatiques afférentsqui arrivent au niveau de la capsule et au niveau du hile (partie incurvée), il y a d’autres vaisseaux lymphatiques qui sortent et souvent il y a du TA, des artères et des veines à ce niveau. Du coup on a le TA, le parenchyme est complètement envahi, on voit un peu la capsule, puis on verra de temps en temps quelques follicules lymphoïdes.
La taille tumorale : facteur de mauvais pronosticLa survie est plus ou moins mauvaise en fonction du stade histologique.-> Quand c’est NO = 75%-> Quand c’est N>0 = ¼ des patients quand les ganglions sont positifs.
Survie globale à 5 ans :→ T < 20mm 91 %→ 20mm < T < 50mm 80 %→ T > 50mm 60 %
On a différents types de progressions :- survie globale : tout événement confondu, que la patiente est re progressé, récidivé ou pas.- survie sans progression = PFS (Progression Free Survival) en absence de tout signe clinique de la maladie pendant la période considérée. C’est encore mieux ça.
Edition 2017 du PTNM des cancers du sein :Classe les tumeurs en fonction du fait que la tumeur sont in situ, qu’elle soit d’une taille allant de 0-1cm à 2cm ou voir même ici plus de 5cm ou qu’il y ait une extension thoracique à la peau ou une induration sous cutanée, etc.. (pas apprendre !)
On voit sur cette diapo (à ne pas apprendre) qu'on a des degrés de micro-invasion et d'invasion locale plus ou moins gradués selon la profondeur de l'invasion dans le chorion, dans les tissus adjacents, etc... C'est aussi un facteur de mauvais pronostique. En fait plus le stade est élevé au niveau anapath plus c'est mauvais pour le patient.
pas apr
N : en fonction du nombre de ganglion analysé, il en faut plus de 6 N0+ : quand y aura juste qqs cellulesN1 : micrométastases avec 1à 3 ganglions etc .. -> tout ceci est l’affaire des spécialistes et c’est révisé en fonction des courbes de survies et de l’évolution des traitements.
On a aussi un tableau pour les ganglions. Si on a analysé moins de 6 ganglions en pathologie, on dit que ce sera non-contributif donc pNx. Il faut en analyser plus de 6 pour que ça soit contributif. Par contre on a pu en analyser plein au niveau c, mais ce n'est pas la même résolution.pN0 c'est quand on en a analysé au moins 6, mais qu'il n'y en a aucun d'envahi. Ensuite pN0(i+), quand il y un micro-amas de cellules tumorales qui se voit sur coupe histologique.
Il faut tenir compte des facteurs de pronostics en fonction des traitements qui vont être reçu. Comme c’est pour un traitement standard, le traitement standard évolue, il va donc falloir refaire le PTNM en fonction de tel ou tel traitement de référence.
Les facteurs pronostics : on estime en fonction du facteur étudié le pronostic, dans un groupe de patient qui reçoit un traitement de référence
Les facteurs prédictifs : on compare un traitement nouveau au traitement de référence et on va voir quels sont les facteurs qui prédisent la réponse à ce nouveau traitement
Le grade histologique ou cytologique :
Le grade histologique du cancer du sein nous montre que tout ça se fait au microscope et va donner à la fois la différenciation architecturale (différenciation tubuloglandulaire -> bon pronostic) de ce cancer mais aussi la différenciation cellulaire : plus il y aura de cellules anormales moins c’est de bon pronostic et plus il y aura de mitose moins c’est de bon pronostic
on aura des grades 1 qui ont une survie à 79% à 5ans des grades 3 qui sont des tumeurs plus agressives, où à peu près 40% des patients vont être décédés à
15ans.Il s’agit d’un grade inventé par monsieur Elston et Ellis. Ce sont des notions qui peuvent également évoluer. Tout ceci c’est l’anapath qui le fait.

C’est l’aspect des cellules qui va être complété parfois par des immuno marquages, le fait que ça exprime ou pas les kératines, qui va compléter le tableau concernant les éléments de différenciation ou pas. C’est le fait que le grade (pas tout à fait le même dans les différents cancers) ici pour le cancer du sein, c’est la présence d’une différenciation tubulo-glandulaire. Cad que les cellules gardent un aspect glandulaire ou d’un canal excréteur. Donc plus elles sont proches des cellules glandulaires acineuses ou de canal excréteur normal, plus elles vont être un grade bas, cad grade 1.Ensuite les aspects cytologiques au sens large, cad la morphologie noyau. Plus le noyau va être atypique, plus le rapport nucléocytoplasmique va être élevé. Plus le noyau va être bizarroïde, nucléolé avec des morphologies un peu lobulées, plus ça va augmenter le grade.Et puis enfin, l’index mitotique (= ratio de 0 à 1, 10% de cellules en mitose = 1 sur 10, 1 ça veut dire que toutes les cellules sont en mitoses ce qui n’arrive pas, même dans les lignées cellulaires in vitro qui se divisent à toute vitesse, l’index mitotique n’est pas de 100%), les anapath les comptent au fort grossissement en regardant si ils voient des cellules en métaphase, anaphase, télophase et puis ils vont compter. Ils vont compter n cellules et le pourcentage. Ou alors ils font des marquages au PCNA (Proliferating Cell Nuclear Antigen) ou au Ki-67. Plus le pourcentage est élevé, plus l’index mitotique est élevé.
-> Du coup le panachage de ces 3 critères va donner les tumeurs de grade 1, 2 ou 3 selon qu’on ait 0, 1, 2, ou 3 caractères atypiques sur les cellules.- Grade 1 = cellules bien différenciées, elles ont peu d’atypie nucléaire et les index mitotiques sont faibles.- Grade 2 = on commence à avoir une cellule un peu moins différenciée et/ou des polymorphismes nucléaires et/ou un index mitotique.- Grade 3 = il y a au moins 2 ou 3 des critères anormaux.
Le type d’origine cellulaire très important
C’est arrivé depuis environ une dizaine d’année. On distingue des cellules luminales A, des cellules luminales B, des cellules basales. On s’est aperçu qu’en fonction de la différenciation du cancer, s’il est luminale A ou B, ou basales, il a un pronostic différent.Quand on fait un profil luminale A ou B ou Basale on a des courbes de survie en mois, ici c’est la survie globale, qui peuvent s’effondrer notamment pour le type basale et pour un autre type de cancer, le cancer R2 positif.Le profil de différenciation des cellules cancéreuses va expliquer les différences de pronostic.Les techniques de profiling du cancer du sein sont particulièrement à la mode actuellement. Pour la petite histoire, ce test coûte 1800euros par patient, donc le gouvernement veut le mettre en place mais n’a pas

l’argent. Du coup les tumeurs étaient envoyées à l’institut Curie, mais dès le mois de mai l’enveloppe globale française a été épuisée, donc on ne pouvait plus avoir les test à moins que les patients payent de leur poche.
Pourquoi ces tests sont importants ?Car quand on a un luminal A, on a un meilleur pronostic et surtout pour les luminaux A, on ne fait pas de chimiothérapie adjuvante. On fait donc faire des économies à la sécurité sociale.Il a été démontré aussi récemment qu’on surtraitait les patientes porteuses des cancers du sein à cause de cette hantise de voir apparaitre des métastases. Mais, comme on n’avait pas de marqueurs, on surtraitait tout le monde.Ce qui est maintenant connu c’est qu’à cause du type d’origine cellulaire, les cellules luminales A, qui sont en plus réceptrices aux œstrogènes, et progestérone positive, ont un meilleur pronostic que les cellules basales qui sont des triples négatives pour les récepteurs à l’œstrogène, à la progestérone et pour R2.On arrive donc déjà à ces résultats avec l’immunohistochimie. Mais, il y a aussi le test de profiling des tumeurs mammaires qu’on peut faire, où à partir d’un bloc en paraffine, on peut faire de la prédiction de groupe d’origine cellulaire, ce qui va permettre de décider ou non d’une chimiothérapie adjuvante.
Facteurs histologiques prédictifs dans le cancer du sein.
Petit topo :- Quand elles expriment les récepteurs : elles sont luminale A- Quand elles sont triples négatives : elles sont basales- Quand elles sont Rp négative et HER2 négative : elles sont basales- Quand elles sont RERP positive et souvent R2 positive : elles sont R2, Her2
Il y a donc plusieurs paramètres et ça devient donc plus ici une affaire de spécialistes.
Les récepteurs hormonaux c’est à la fois des facteurs et le récepteur R2. C’est à la fois des facteurs pronostics car quand les cellules sont positives pour les récepteurs hormonaux RERP, on va dire qu’elles sont luminale A. Quand elles sont triple négatives elles sont basales. Quand elles sont R2+ elles sont Her2.
Ces récepteurs hormonaux sont à la fois des facteurs pronostics mais également des facteurs prédictifs . Cad que quand on a des récepteurs hormonaux, on est bons répondeurs à des thérapeutiques antihormonale, qui vont dépriver la tumeur de la stimulation hormonale.

- On donne aux cellules qui sont positives aux récepteurs aux œstrogènes des anti aromatases ou des anti œstrogène.
- On va donner aux cellules qui sont R2 positives un anticorps antiR2- Et quand les cellules sont en cycle, on va plutôt leur donner une chimiothérapie
Tout ça est évalué par immunohistochimie et si besoin en FISH.
Cela est très important pour la thérapeutique, se sont des facteurs qui vont rentrer en compte, qui peuvent être pronostics mais qui sont prédictifs aussi de la réponse thérapeutique.Ce qu'on veut voir c'est la sensibilité des cellules tumorales dans le cancer du sein. On va regarder l'expression des récepteurs hormonaux (progestérones, œstrogènes), cela va pouvoir donner un certain nombre de traitements. Il y a plusieurs possibilités thérapeutiques, par exemple les inhibiteurs de l’aromatase qui est l’enzyme de conversion qui permet de transformer le cholestérol en hormone stéroïdienne et donc chez la femme on peut bloquer la synthèse des oestrogènes. Même si la cellule exprime le Rc et qu’il y a plus de ligand, ça ne va pas avoir le même impact et comme ces cellules sont oestrogénodépendantes, on va diminuer la prolifération de ces cellules. Ca va pas tuer les cellules nécessairement mais ca va au moins avoir un effet cytostatique, ça va réduire la prolifération des cellules voir induire l’apoptose quand on bloque cette voie.Une autre possibilité, c’est un traitement par des antagonistes qui vont entrer en compétition au niveau du Rc. Ce sont des Rc cytoplasmiques qui sont à destinée nucléaire parce quand ils sont activés par les stéréoïdes, ils sont transloqués dans le noyau où ils vont activer la transcription, c’est des molécules qui jouent le rôle de facteurs de transcription stéroïdes dépendant. Donc se sont des hormones lipophiles qui diffusent bien à travers la membrane. Si on arrive à avoir des médicaments qui ont une affinité pour le Rc qui diffuse bien à travers la membrane plasmique des cellules mais qui n’ont pas d’effet agoniste sur ce Rc, alors on peut bloquer l’action faite de ces Rc. Parfois, c’est une action combinée d’antagonistes avec aussi les inhibiteurs de l’aromatase, comme ça, on bloque les stéréoïdes endogènes, on bloque les Rc, on fait des fois des actions combinées.Ces molécules ne sont pas parfois des antagonistes complets, elles peuvent avoir encore pour certaines d’entre elles des effets d’agonistes partiels. Mais globalement ces médicaments, Tamoxifène, Ramoxifène, ont un effet antagoniste sur la prolifération des cellules épithéliales mammaires.
Erb2 (= Her2) est un récepteur à activité tyrosine kinase de la famille de l’EGFR (= RB1) exprimé dans certaines situations comme dans le cancer du sein où il y a des amplifications géniques qui aboutissent à la surexpression de la protéine. Donc la, il y a une thérapie ciblée qui est un Ac bloquant, un peu comme lecetuximab et le critère d’indication cette fois ci est l’expression du Rc car on sait que quand il est

surexprimé, on sait qu’il y a un bénéfice thérapeutique. Donc là on peut regarder comme bio marqueur en première intention, l’expression du Rc et puis éventuellement on fera de la FISH pour confirmer.Et le dernier facteur prédictif, c’est les anti-mitotiques, se sont des molécules de chimiothérapies qui agissent comme anti-mitotique donc des cytostatiques ou des cytotoxiques, soit le Taxol qui agit sur les fuseaux mitotiques soit les sels de platine qui ont des effets sur les cellules en cours de division car ça induire des dommages de l’ADN qui ne seront pas réparés et du coup ça va induire l’apoptose des cellules. Tous ces traitements vont mieux marcher si les cellules se divisent beaucoup.
Il a horreur des termes anapath car ils vont parler de détection positive et détection négative (= absence de détection). La détection positive est difficile car parfois on ne voit pas de marquage mais là on a pas vraiment de contrôle positif contrairement à ce qu’on avait pour la E-cadhérine, pour les molécules du mismatch repair. Les cellules normales peuvent éventuellement exprimer un petit peu plus ou moins, etc mais c’est plus difficile parfois l’interprétation. Ce sont des Rc qui sont à l’état non activé, ils sont cytoplasmiques ou nucléaires.Autres facteurs histo pronostiques dans le cancer du sein : type d’origine cell coure de survie / embole vasculaire
On voit qu'ici c'est positif, c'est unmarqueur nucléaire (en effet œstrogène = stéroïde = lipophile donc atteignent le noyau). En fait les récepteurs hormonaux des stéroïdes sont cytosoliques ; mais quand ils sont activés, il se dimérisent et vont dans le noyau pour réguler la transcription.


Alors que ci-dessous, on voit une absence de détection ici dans le tissu tumoral mammaire, avec quelques cellules qui peuvent être positives sur la lame et qui pourront servir de contrôle.
On a là une valeur à la fois pronostique et prédictive.
- Prédictif : quand c'est récepteur hormonal positif, on va traiter avec du Tamoxifène et des inhibiteurs d'aromatase car la patiente exprime ces Rc et donc on peut suspecter que les cellules sont homorno dépendante. Donc si on bloque cette voie, on va avoir un impact thérapeutique
bénéfique On va alors bloquer le récepteur et bloquer la production endogène d'œstradiol → effet thérapeutique, la courbe remonte vers le haut.- Mais aussi Pronostique : au moins au début de la maladie, c'est un facteur pronostique positif. On a une valeur pronostic positive puis après on a le cross over cad que les courbes se croisent et à la fin les tumeurs qui persistent pendant longtemps, finissent par être plus agressives car il y a des mécanismes d’échappement, etc. En plus de ça, elles vont bénéficier d’un traitement qui va tirer la courbe vers le haut. C’est un facteur de bon pronostic et un facteur en faveur de l’immunothérapie, en revanche les tumeurs qui sont estrogènes Rc négatives, elles répondent mieux à la chimiothérapie.Chaque type de maladie selon ses mécanismes oncogéniques ne va pas répondre au même traitement.
- Tumeurs RE+ répondent à l'hormonothérapie- Tumeurs RE- ne répondent pas à l'hormonothérapie.
En ce sens vous voyez que les patientes RE+ ont de meilleurs chances de survie contrairement aux RE-, certainement dû à des facteurs biologiques conjoints qui font que ces tumeurs ont une étiologie moléculaire moins agressive.A la fin par contre c'est un facteur de mauvais pronostique, car les cellules sont en phase d'échappement et perdent leur hormono-dépendance. Les cellules tumorales acquièrent des mutations qui leur confèrent une agressivité supplémentaire.
Donc cela va conditionner le traitement, si le Rc oestrogénique est positif, on peut donner du Tamoxifène,
Le récepteur R2 qui est un récepteur de type tyrosine kinase, c’est le deuxième membre de la famille EGFR, c’est EGFR2. Lorsqu’il y a un facteur de croissance qui arrive au niveau du récepteur, il va y avoir effectivement une dimérisation, et donc une activité des tyrosines kinase, ce qui va entrainer une signalisation.Ce récepteur, dans le cancer du sein, peut être exprimé par un phénomène d’amplification génique, qui s’observe dans environ 10% des cancers du sein. Donc ceci va définir le groupe R2elmich, un groupe particulier de tumeur du sein, qui comme on l’a vu tout à l’heure sur les courbes de survie pronostic, avait plutôt un mauvais pronostic par rapport aux cellules luminales A ou B. C’est donc plutôt un cancer du sein agressif par son pronostic spontané. Si vous soumettez ce cancer du sein à un traitement standard comme les autres, la courbe de survie s’effondre, donc il s’agit plutôt ici de tumeurs agressives.Cette amplification de R2, qui joue un rôle dans l’oncogénèse de cette tumeur va entrainer beaucoup de transcrit de R2 et beaucoup de protéine de R2 à la surface des cellules tumorales.La première technique qu’on va pouvoir utiliser pour dépister ces tumeurs R2 positive c’est l’immunohistochimie.
Donc valeur pronostique variable dans le temps :Leur présence → bon pronostique à court terme mais mauvais à long terme.Leur absence → mauvais pronostique à court terme (< 6 ans) mais bon pronostique à long terme.
Enfin, ErbB2 est un récepteur de la famille tyrosine kinase. Il a l'habitude de se dimériser avec un autre partenaire de la famille comme ErbB3 → hétéro-dimère.On ne connaît cependant pas le ligand direct de ErbB2, on appelle ça un récepteur « orphelin ». Quand un ligand se lie par exemple sur ErbB3, il va être activé, va se dimériser et se transphoryler avec ErbB2. Donc s’il y a un cancer, il y aura amplification du gène codant pour le récepteur, donc surproduction d'ARN, de la protéine et donc amplification de la voie intracellulaire en aval.On a une amplification de ce gène dans 1/10ème et 1/5ème des cancers du sein. On va avoir une augmentation du nombre de copies du gène, ça on peut le voir en IHC, c’est indirect car si le nombre de copies du gène est augmenté, la transcription va être augmentée et donc la traduction va être augmentée aussi.La voie de transduction en aval aura pour effet de provoquer l'hypertrophie des cellules, mais elle aura aussi un rôle mitotique pour la synthèse d'ADN et métastatique. En effet, si on introduit expérimentalement plusieurs amplifications de ce gène in-vitro dans des cellules, on pourra observer une perte d'adhésion, une augmentation de la capacité protéolytique des cellules, etc...Le gène est sur le chromosome 17.
On peut distinguer ici en première intention, en immunohistochimie les tissus normaux qui sont négatifs (structures canalaires, lobulaires normales en témoin) et puis on voit la tumeur avec un fort marquage membranaire pour le récepteur tyrosine kinase. Quelque soit l'origine de cette surexpression, on va traiter à l'herceptin.
Les laboratoires utilisent des scores pour graduer la coloration et communiquer entres eux. Ici donc c'est très positif 3+. Dans ce cas là on ne fait même d'hybridation in situ, étant donné que la protéine est surproduite. La cause pourrait être une amplification génique, ou encore un facteur de transcription qui est suractivé par phosphorylation ou suractivé donc amplification de la protéine récepteur tyrosine kinase sans altération du gêne.
Quelque soit l'étiologie, on utilisera l'herceptin si positivité aux ErbB-2 3+. Cependant si c'est négatif ou 1+ on ne fait même pas de FISH ; si on est ErbB-2 2+ donc score positif intermédiaire on va devoir absolument faire une FISH pour vérifier l'amplification génique.
Du coup ici, on va faire une FISH (toujours en 2 couleurs) on aura une sonde centromérique« contrôle » en vert et une sonde du locus ErbB-2 en rouge. Donc si c’est normal, on aura 2 spots rouges et 2 spots verts. Si il y a des polysomie, il y aura l’augmentation des signaux rouges et des signaux verts. Si on a une amplification génique, il y aura un déséquilibre entre les rouges et les verts. Donc on peut distinguer entre polysomie et amplification et du coup il y a un cut off, c’est quand il y a un déséquilibre de ratio entre les nombres de signaux rouges et de signaux verts qu’on peut dire qu’il y a une amplification génique. On contre colore au DAPI tout le temps parce que sinon on ne voit pas les limites des cellules c’est sur fond noir. On colore au DAPI, car il permet de limiter les noyaux, comme ça quand on compte les signaux, on compte les signaux correspondant à un noyau. Le problème du comptage de signal en FISH, c’est pour ça qu’on préfère faire des empreintes pour un certain nombre de techniques parce que les empreintes on a des cellules intactes qui se sont déposées sur la lame. En coupe histologique, on peut avoir des coupes qui peuvent superposer des cellules ou couper des cellules en deux. Donc à ce moment là, il faut compter une centaine de cellules pour être sur que l’augmentation des signaux ne sont pas des superpositions et que des diminutions de signaux c’est des vrai délétions et ce ne sont pas des cellules coupées en deux ce qui fait qu’on perd des signaux.

L’immunohistochimie va nous permettre de définir qu’une tumeur du cancer du sein est R2 positive.Vous avez à droite, une tumeur qui a un score 3+ avec un marquage fort dans le cadre de toutes les cellules tumoralesVous avez en bas à droite, une cellule d’une tumeur qui est de score faible 1 voir 0 si il n’y avait pas du tout de marquageSur la partie droite de la diapositive, on voit en haut, une patiente qui va recevoir le trastuzumab qui est un anticorps antiR2 (qui coûte 30à 40 mille euros par an à la sécurité sociale mais à ce prix on va au moins savoir pourquoi et ça va marcher, si ça va traiter voir même guérir cette tumeur qui est sensible à l’herceptin)Alors que la maladie, en bas à droite, qui n’exprime pas R2, ne répondra pas à ce traitement et on ne va pas lui faire de RCP.Il peut y avoir aussi des situations intermédiaires avec un score deux plus. Dans ces cas là, on demande à faire de la FISH. Cad qu’on va faire une sonde centromérique du chromosome 17 et une sonde de R2 sur le 17q et on va essayer de voir le nombre de copie entre le centromère et le gène R2.
Quand on va avoir un gros paquet de rouge, ça veut dire qu’il y a une amplification de R2. Quand on a un ratio de deux deux ça veut dire qu’il n’y a pas d’amplification. Quand on a une situation intermédiaire, on va faire le ratio du nombre de spot rouge sur le nombre
de spot vert.Par exemple si le ratio est de 1,45 ça veut dire qu’il n’y a pas un ratio supérieur à 2 on ne donnera pas d’herceptin.L’herceptin on va les donner :
- chez des patients qui sont 3 croix, pas besoin de faire la FISH- ou qui sont deux croix (parce qu’on a fait la FISH avec un ratio supérieur à 2 de R2 sur le
centromère)
Donc ça va nous permettre de non seulement faire des économies au niveau de la sécurité sociale, mais surtout ça va nous permettre de proposer un autre traitement aux patients qui sont R2 négatif. C’est donc très important de voir que dans ces terrains assez ciblés, on a un double intérêt. Un intérêt de sélection économique des patients, mais surtout de toujours donner le traitement le mieux adapté à la tumeur de la patiente.C’est aussi important de bien définir qu’elles sont les tumeurs R2 amplifiée par rapport au non amplifiée, puisqu’on a vu que dans le profiling, la survie change en fonction du groupe, il ne faut donc pas se tromper de groupe de patient.
Donc on fait les screening en IHC et en première intention on fait la technique la plus simple mais il peut y

avoir des faux négatifs, puisque sur les cellules normales il n’y a pas un marquage très fort de Rb2 (protéines qui sont très régulées et pas exprimées à fort niveau à l’état physiologique sur des cellules différenciées comme ça, du coup on a pas de contrôle interne). Donc si c’est négatif la lame, ça peut être aussi un problème de lame, c’est pas forcément histologique, pré-analytique, etc.Mais quand c’est très positif, on a pas d’ambiguïté, et là on ne fait même pas la FISH, quand ça « crache » fort, on voit un beau marquage membranaire en immunopéroxydase sur les cellules tumorales, donc on sait que c’est pas un faux positif, parce qu’on a des cellules qui sont négatives sur les cellules normales et donc on peut aller direct avec l’herceptin, on n’est pas obligé de faire un examen moléculaire complémentaire car on s’en fiche que la surexpression soit liée à une amplification ou à un mécanisme épigénétique, dans ce cas là, il y a une surexpression de la cible. A priori les cellules dépendent pour leur voie de prolifération de cette cible.
Quand on a un score faible comme ça intermédiaire des cellules, on peut se poser la question si c’est parce que le marquage n’a pas bien marché ou au contraire parce que l’expression est faible et donc à ce moment là on fait une FISH pour confirmer. Si la FISH montre un résultat d’amplification génique, à ce moment là on traite par l’herceptin car on peut se dire que le faible marquage est lié à un problème de la qualité de la lame. En fait les acides nucléiques et les protéines ne réagissent pas forcément de la même façon aux problèmes de fixation et tout. Donc on peut avoir un impact sur les protéines alors que l’ADN va lui même être bien conservé. L’ADN est assez costaud donc si on prend un
prélèvement et qu’on ne le fixe pas tout de suite, ce n’est pas l’ADN qui va se dégrader en premier mais l’ARN et les protéines qui sont assez sensibles. Donc on peut avoir un problème qui va impacter les protéines techniques du prélèvement et pas l’ADN, donc on va détecter la FISH positive et donc que c’est in vivo l’expression de la protéine sera présente si le gène est amplifié.
Quand c’est négatif, on ne fait pas de FISH et on ne donne pas le médicament. Ce qui peut perdre certains patients parce qu’en fait on peut avoir une absence de détection et plus ou pas de marquage et que ça soit un problème pré-analytique (lame pas de bonne qualité) et on va même pas vérifier en FISH mais c’est comme ça les recommandations. C’est dommage car il y a peut-être un petit pourcentage assez faible de patientes qui vont pas recevoir l’herceptin alors que peut être elles étaient positives en FISH mais que leur lame d’histo était mauvaise et qu’on a pas vu le marquage de Rb2.Le prof pense que la pathologie moléculaire est plus fiable que l’IHC dans un certain nombre de cas et donc il ne comprend pas pourquoi ils ne feraient pas la FISH ici.

Vous voyez ici, sur l'image du haut, cellules normales enfin du moins pour le locus ErbB-2 / centromère avec pour chaque cellule 2 signaux verts et 2 signaux rouges donc aucune amplification de ErbB-2. Cependant ces cellules sont peut-être tumorales et présenteront peut- être une amplification à un autre oncogène. Pour l’image en dessous on voit bien qu'il y a une amplification génique du gène ErbB-2 pour ces cellules anormales → majorité de signaux rouges.
Quand on a que 2, 3 copies surnuméraires ce sont des gains et quand on en a beaucoup on parle d’amplification.
Si les nombres de signaux rouges et verts sont les mêmes, on parle de polysomie. Il peut y avoir plusieurs types de polysomie, les cellules sont polyploïdes, ou polysomies soit plusieurs copies du même chromosome.
L’intérêt pronostic de R2, en dehors de la thérapeutique, par rapport à un traitement de référence standard, est de voir que ces patients ont spontanément un plus mauvais pronostic que les autres qui sont R2-. Mais par contre si on les traite avec l’herceptin, à ce moment là, le traitement (pour les patient R2+ avec un traitement à l’herceptin) aura un bénéfice important, on voit donc que la courbe de survie n’est pas la même
Ici on a deux types d’impacts qui ne sont pas forcément dans le même sens. La présence, l’expression et l’amplification de Rb2 est un facteur sur le plan pronostic qui est mauvais. Tout à l’heure, l’estrogène Rc était un facteur pronostic et prédictif de bonne augure car il répondait à la thérapie anti-hormonale.
Ici c’est un facteur pronostic mauvais car les cellules qui sur-expriment Rb2 ont un impact sur la

prolifération cellulaire mais aussi sur les capacités migratoires et métastatiques des cellules. Si on prend une lignée cellulaire du cancer du sein et qu’on transfert un vecteur viral qui code pour Rb2 pour faire sur- exprimer artificiellement, pour faire comme ci c’est une tumeur qui exprimait Rb2 mais on la crée artificiellement, on va augmenter le processus des capacités migratoires et métastatiques de ce cancer dans des systèmes in vitro et dans des modèles expérimentaux chez l’animal. C’est un facteur de mauvais pronostic mais c’est un facteur prédictif qui va permettre quand on traite avec l’herceptin de faire remonter la courbe de manière plus favorable.
On voit ici une mammographie qui a l’air calcifiée, hypodense en échographie. On prend la pièce d’exérèse, on fait l’histologie et on voit que c’est un carcinome.
Le cas clinique : Une femme qui, à une mammographie, révèle un nodule de 20mm au niveau du sein gauche. On peut éventuellement faire soit ce qu’on appelle une microbiopsie avant l’opération pour avoir un diagnostic. Soit on décide d’opérer d’emblée.
On fait donc une exérèse, une tumorectomie → obtention d'une tumeur assez grosse pas très bien limité donc ressemble à un carcinome infiltrant. Il y a un filtrant au niveau ganglionnaire.
On voit ici une tumeur de 22mm, blanchâtre, mal limitée qui évoque en premier lieu un cancer. On trouve un carcinome canalaire infiltrant peu différencié avec métastases, en plus dans un ganglion axiliaire. Vous avez le tissu lymphoïde en périphérie et toutes les cellules cancéreuses. On voit également des gros noyaux avec un grade relativement élevé pour l’agressivité de la tumeur

Une immunohistochimie nous montre des RE + et des RP+, une prolifération et surtout un score 3croix pour le gène R2.Cette malade rentre donc dans le groupe des cancers R2 positifs, qui vont recevoir le stratus humainElle était à PT2N, ce qui va être ganglion envahit A
- On va lui faire à cause des récepteurs hormonaux une anti hormonaux thérapie ou hormonaux thérapie.
- On va lui faire à cause de R2 une chimiothérapie combinée de herceptin.- Et comme elle avait des ganglions on pourra lui faire une radiothérapie du sein et des aires
complémentaires.En matière de cancer mammaire, l’enjeu c’est d’identifier, les patients qui sont avec ces cancers agressifs. Quand on est dans ce groupe là, on dégaine toute l’arsenal thérapeutique.Si on pouvait avoir l’arme nucléaire contre ces cancers, on utiliserait l’arme nucléaire, parce que on sait que si globalement on ne fait rien, il ne restera plus que 20% des patientes à 5ans.Donc en faisant ça, on fait probablement un traitement difficile à supporter pour les patientes, mais il s’agit d’un traitement qui peut sauver un certain nombre de patientes.A l’inverse, il y a des cancers de plus faible grade pronostic, qui sont des cancers luminaux A, avec une survie à 5ans voir même à 10ans qui est relativement satisfaisante.Tout le problème actuellement est d’identifier celles qui vont avoir besoin d’une chimiothérapie par rapport à celles qui n’ont pas besoin d’une chimiothérapie, c’est là que les techniques de profiling peuvent peut être aider.
On voit que c’est positif aux Rc hormonaux, on voit qu’il y a un index mitotique élevé et que c’est positif pour le Rc Rb2. Même si sur le plan clinique il y a une invasion ganglionnaire, on a ici un cas qui n’est pas le pire puisqu’on a un arsenal thérapeutique assez complet chez cette patiente. On peut donner les anti- mitotiques, la thérapie hormonale, l’herceptin. On peut faire une thérapie combinée qui va maximiser les chances de faire régresser sa maladie. On a donc un pronostic plutôt favorable.
Conclusion Cas clinique :
1. Diagnostic
Carcinome canalaire infiltrant de 22mm
Grade Histopronostique III (différentiation 3, anisocaryose 2, mitoses 3)

Absence d’emboles vasculaires péritumoraux
Une métastase ganglionnaire axillaire sur 12 ganglions prélevés = Stade pT2N1a
Présence de récepteurs hormonaux ( IHC)
Présence d’une surexpression forte 3+ de Her2( IHC)
Index de prolifération Mib1 à 40% (IHC)
2. Traitement
– Radiothérapie du sein et des aires ganglionnaires sus clavicualire et mammaire interne
– Chimiothérapie combinée à l’Herceptin
– Hormonothérapie
III. Cancer pulmonaire.
Intérêt du dossier : Diagnostic des adénocarcinomes pulmonairesIllustration des facteurs pronostiques et prédictifs de la réponse thérapeutique (thérapeutiques ciblées)
Pour nous donner un ordre d’idée :Dans le labo dans lequel Merlio se trouve, on analyse 1700 cancers du poumon par an. Ce n’est pas 1700 nouveaux cas, c’est 1700 cancers pulmonaires métastasiques, il y en a d’autres qui ne sont métastasiques. Globalement le volume cible pour les cas, c’est un cancer métastasiques ou avancé 20milles. Actuellement, on en compte 15milles par le système mis en place, ce qui est déjà pas mal. Ce qui veut dire que trois quart des patients accèdent à un diagnostic moléculaire, ce qui est remarquable pour la France par rapport aux autre pays.A la Réunion, il y a à peu près 180 patients par an qui sont testés pour ces anomalies génétiques.
1) Observation clinique. - Mme Françoise, 43 ans, non fumeuse.
- Toux chronique, amaigrissement de 5 kg en 3mois,- Examen clinique normal.- Radiographie thoracique : opacité douteuse au niveau du poumon droit.
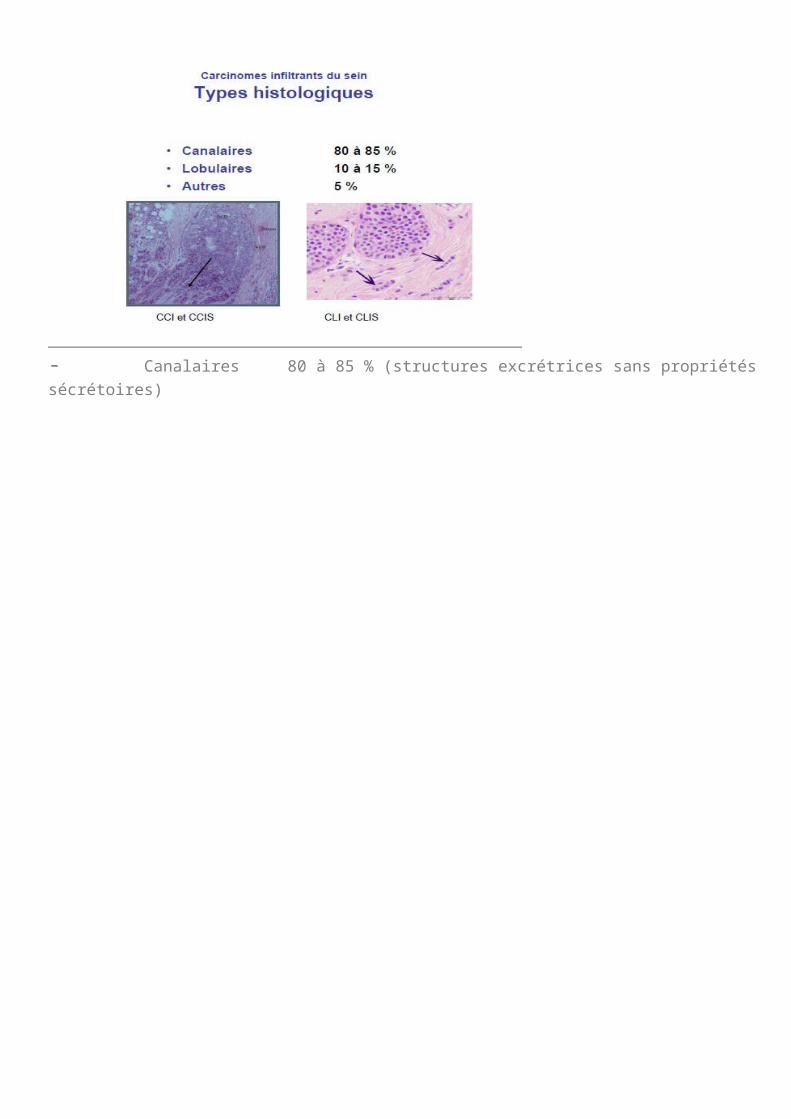
Si on fume, on favorise le cancer du poumon. Si on ne fume pas, on peut quand même malheureusement en avoir un aussi. Cela est probablement lié à des subtilités à la fois individuel mais aussi à la pollution. On sait que les asiatiques font beaucoup de cancers du poumon, parce que non seulement ils fument, mais en plus c’est pollué chez eux, donc il y a énormément de cancer du poumon en Chine.
Toux chronique, amaigrissement en 3mois. Qqn qui a une toux qui est persistante, il faut surtout l’examiner et faire une radio thoracique. La radio thoracique ne coute pas cher et ça permet de dépister un certain nombre de cancer, lorsqu’il existe une image comme par exemple celle-ci.
Cette image ici est périphérique, on a un nodule parenchymateux tumoral, qui selon les radiologues correspond à ce que l’on appelle une opacité en verre dépoli.Dans ce type de cancer, qui est situé en plein parenchyme pulmonaire. Ce qu’il faut également comprendre, c’est qu’on est dans le parenchyme pulmonaire, près des petites bronches et des alvéoles. Pour aller chercher ce type de cancer, on va plutôt passer par l’extérieur, on va faire une ponction biopsie transthoracique. Le radiologue va aller sous scanne, introduire une carotte, c’est un genre de système de pistolet. Il tire sur la tumeur et le pistolet va aller cisailler comme une espèce de baïonnette, comme un forage. Il va aller introduire le pistolet et à un moment il y a un petit ressort, il tire, ça fait une baïonnette et il peut ensuite retirer une autre fois. Le radiologue ramène ainsi de très jolies carottes, qui sont pleines de matériels tumorales. Ce qui permet de faire de l’histologie et donc un diagnostic de ce matériel tumoral.Si la tumeur était au contact du médiastin et au contact des bronches (proches d’une grosse bronche sur l’image), on aurait un autre abord de cette tumeur, qui serait d’aller faire une endoscopie bronchique et une biopsie transbronchique. Cad qu’on introduit un endoscope et qu’on peut à travers la bronche prélever la tumeur qui est en plein parenchyme.Donc très important, il a deux voies d’abord pour faire le diagnostic du cancer du poumon : La voie transthoracique ou la voie transbronchique.
On voit l'opacité en verre dépoli sur la radiographie du poumon. C’est suspect donc on va faire une biopsie trans thoracique sous TDM. On va aller ponctionner et faire une aspiration du tissu pour faire l’examen histologique.
Non fumeuse → c'est une étiologie rattachée plutôt aux adénocarcinomes. Les fumeurs développent des

cancers du poumon généralement différent de ceux des non fumeurs.
3. Examen histologique
On a donc obtenu le matériel tissulaire et ce que l’on observe c’est une prolifération de cellules cohésives formant des lumières, cad des glandes.->On va donc répondre adénocarcinome. Puisque c’est une prolifération tubuloglandulaire.Le problème de cet adénocarcinome, qui est obtenu par ponction sous scanner, est de savoir si cet adénocarcinome est d’origine à proprement parler pulmonaire par rapport à une métastase. Car ça pourrait très bien être une métastase pulmonaire d’un cancer du côlon.
On a une petite carotte de tissu qu’on va examiner et donc on voit une prolifération d’aspect tubulo- glandulaire au niveau du parenchyme pulmonaire. Petit rappel : l’épithélium de type respiratoire est prismatique cilié, on voit des cellules caliciformes, des cellules glandulaires endocrines (voit pas très bien) et puis au niveau de l’épithélium du parenchyme alvéolaire, on a des pneumocytes qui ont un aspect pavimenteux. Donc en fonction de l’histologie qu’on va avoir à certains endroits, on dira que là les cellules on un aspect tubulaire avec des cellules prismatiques, donc elles font plutôt parties d’un épithélium de type respiratoire, pas d’épithélium alvéolaire, se ne sont pas des cellules pavimenteuses.
QCM 1 : Devant ce diagnostic d'adénocarcinome sur le prélèvement réalisé par ponction sous scanner, quelle est la question qui se pose ?
1. bilan d'extension de l'ADK2. mutation de l'EGFR ?3. caractère primitif ou métastatique de l'ADK ?Quel moyen pour résoudre cette question ?
Analyse Immunohisto chimique (IHC)On ne fait pas tout de suite la mutation EGFR car elle n'est présente que dans les tumeurs pulmonaires
primitives, donc il faut d'abord déterminer le caractère de la tumeur.De toute façon, c'est la routine histologique. L'anatomo-pathologiste va donc faire une immunohistochimie que voici : 1H11 .06

Comment éliminer le faite que ce ne soit pas une métastase pulmonaire d’un cancer du côlon ?Il y a cette image, mais le problème est de savoir si c’est vraiment un cancer pulmonaire ou si c’est un cancer du côlon. Car ce n’est pas marqué sur la cellule, si elle est d’origine pulmonaire ou d’origine digestive.
Réponse : l’immunohistochimieEn effet, on va faire de l’immunohistochimie. On va devoir déterminer si c’est primitif pulmonaire ou métastasique.On va donc faire les fameuses cytokératines. En fonction du profil, CK7 ou CK20, ça va être plutôt d’origine pulmonaire primitive, notamment si c’est 7+ et 20-. Et puis on va faire CK7et TTF1.TTF1 c’est un facteur de transcription, c’est le thyroïde transcription facteur one, qui est positif dans les cellules thyroïdiennes (les fameux thyréocytes dans les glandes endocrines) et dans les cellules d’origine pulmonaire.L’ébauche embryonnaire vient de l’endoblaste. Les thyroïdes se différencient à partir de la même ébauche primitive que le poumon. C’est pour ça qu’au stade adulte, ces deux types de cellules expriment les mêmes facteurs de transcription, ce qui reflète leur origine embryologique commune.Si on prend des cellules pancréatiques qui viennent d’une autre partie de l’endoblaste ou si on prend des cellules hépatiques, on n’aura pas le TTF1.
Comme on sait que la patiente n’a pas de cancer thyroïdien, (on pourrait se planter si la patiente avait eu un antécédent de carcinome thyroïdien, il faut donc vérifier les ATCD, mais ici pas d’ATCD de carcinome thyroïdien), c’est donc un marqueur d’épithélium pulmonaire et thyroïdien.
On va faire un certain nombre d’immunodétection et on va voir qu’elles sont légèrement positives. C’est un peu brun au niveau nucléaire pour le Thyroid transcription Factor. C’est un marqueur qui est présent dans la glande thyroïde mais qui est présent aussi dans l’épithélium respiratoire. Il est exprimé dans certains épithéliums, notamment dans les cellules pas trop indifférenciées de l’épithélium respiratoire.On a une expression de la cytokératine 7, donc ce sont des cellules qui sont assez différenciées. Elles expriment TTF et la cytokératine. Ici, ce sont des cellules des unités respiratoires qui sont exprimées par les bronchioles et les alvéoles = unités respiratoires terminales. Donc ici, on a une tumeur assez bien différenciée.
Classification morphologique :
Comment classer un cancer bronchopulmonaire ? Classification OMS 2015 Principes• Classification basée sur morphologie et IHC• Hétérogénéité des carcinomes broncho-pulmonaires

->souvent mixtes (50%)• Type histologique fonction du contingent majoritaire• Regrouper les K non invasifs et ceux à invasion minime car même pronostic favorable
Comment classer les cancers pulmonaires ?C’est basé sur la morphologie et l’immunohistochimie. On a souvent des cancers qui sont de type mixte, cad qu’il n’y a pas une seule morphologie univoque. Il y a souvent un mélange de différents composants. Il peut y avoir des composantes solides, adénocarcinomateuses, indifférenciées etc… et comme il y a des mélanges, on va donner quand même pour donner la classification, le type histologique majoritaire. Dans cette classification, on va distinguer les cancers qui ont un pronostic favorable, cad les in situ ou microinvasif qui ont un bon pronostic spontané, par rapport aux autres qui ont un mauvais pronostic.
Selon le type histologique, parfois quand se sont des tumeurs mixtes avec différents contingents, on va mettre le contingent prépondérant qui pèse le plus lourd dans le diagnostic. On va mettre le carnicome mixte avec contingent principal glandulaire. On va avoir un gradime qui est fait en fonction du contingent, en revanche le moins bien différencié cad que le plus grave va être pris en considération. Donc on n’a pas de classification sur le seul phénotype d’IHC car le phénotype d’IHC est variable. Des fois on n’aura pas de marquage de TTF alors que les cellules sont relativement bien différenciées. Donc se sera un marquage mixte.
Comment classer un cancer bronchopulmonaire ?? Classification OMS 2004
Principes :
•Classification morphologique
et IHC
• Hétérogénéité des
carcinomes broncho-
pulmonaires
souvent mixtes (50%)
• Type histologique fonction
du contingent majoritaire
Ici on a un carcinome qui présente de signes de différenciations glandulaires et plus ou moins des signes de

sécrétions de mucine. Les cellules qui se sont transformées ne sont souvent pas les cellules différenciées, ce sont les cellules un peu progénitrices et du coup elles vont se différencier plus ou moins. Ici elles ont pris plutôt une différenciation de cellules prismatiques, mais elles peuvent quand même avoir pris quelques caractéristiques de mucosécrétion sans être caliciforme pour autant. Donc c’est une tumeur relativement bien différenciée qui garde les caractéristiques de l’épithélium normal auquel on se réfère et donc c’est un grade1. Le phénotype primitif est pulmonaire puisque en plus de l’aspect histologique, en immuno il est positif pour cytokératine 7 et TTF1.
Il est passé sur la classification car elle change tout le temps.
Voici la classification des adénocarcinomes qu’on ne nous demande pas d’apprendre. Mais de retenir qu’il y a un groupe de cancer minimum invasif ou préinvasif, qui a 100% de survie à 5ans après réjection. Donc, ce type de patients est opérable d’emblé. Malheureusement à la Réunion, pour des raisons d’accès aux soins mais aussi peut être dû à la consommation tabagique etc, il y a très très peu de patients opérables de ce type de cancer (microinvasif ou avec une survie à 100%). Le plus souvent, ici à la Réunion, il y a des cancers qui sont souvent métastatiques d’emblé ou invasif d’emblé. C’est donc assez rare que l’on ait ici des cancers au stade microinvasif ou au stade préinvasif.

Merlio a été troisième auteur sur une publication de Lancet, qui a été publié il y a presque un an, par l’inter groupe francophone de cancérologie thoracique. Ils ont analysé 1500 cancers de cette cohorte de 14000 cancers analysés par les centres français. Cette publication montre, comme d’autres, que dans les cancers du poumon, on a soit des adénocarcinomes, soit des cancers épidermoïdes squameux (=kératinisant).La biologie de ces cancers, soit l’oncogenèse de ces cancers, est variable, à la fois selon le type histologique et selon les habitudes du patient. Par ex, les patients qui sont fumeurs vont faire des cancers avec des mutations caras ou des cancers épidermoïdes.Les patients qui sont non-fumeur, notamment asiatiques, vont faire plutôt des cancers multiproegfr (c’est 16% de la cohorte). Ici, à la Réunion on est plutôt à 21%, on a fait les statistiques sur 4ans. 21% car il y a une population asiatique qui fait monter la fréquence de l’egfr. Il n’y a pas la même oncogenèse selon que l’on soit fumeur, caucasien, asiatique etc, donc ce n’est pas la même étiologie.
En dehors du fait, de décrire l’oncogenèse de ces cancers (adénocarcinomes, cancers épidermoïdes) ce qui est très intéressant dans cette histoire de mutations génétiques, c’est que depuis une dizaine d’année, des industriels au premier rang desquels (Pfizer, Astrazeneca..) les gros groupes industriels ont mis à chaque fois, un milliard d’euros pour développer des médicaments contre ces principales mutations génétiques.Globalement, le milliard d’euro qui est mis, marche une fois sur deux ou trois ou cinq. Globalement l’industrie pharmaceutique décrite investit aussi. Le retour sur l’investissement, quand le médicament marche, est qu’elle empoche 10 fois ou 20 la mise environ. Donc il y a toute une économie de cet investissement, que le système publique serait incapable de faire, contrairement à ces grands groupes multinationaux.
Diagnostique histologique :Le diagnostic est très complexe en histo pour le poumon. On fait un bilan d’extension qui est négatif, un bilan préopératoire normal. La tumeur est classée sur le plan clinique, T2aNOMO. C’est une patiente qui est éligible pour un traitement radical curatif par chirurgie (lobectomie supérieure droite + curage médiastinal pour voir si il n’y a pas d’envahissement au niveau ganglionnaire).
Adénocarcinome
= carcinome présentant des signes de différenciation glandulaire et/ou des signes de sécrétion de mucine
Bien différencié (grade G1)

Phénotype primitif pulmonaire
(Aspect histologique + IHC : CK7+, TTF1+)
Ce qui est intéressant là dedans, c’est que non seulement il faut faire un diagnostic étiologique mais aussi faire un diagnostic moléculaire pour pouvoir traiter les malades.Ici, on avait un adénocarcinome bien différencié primitif pulmonaire TTF1+ , CK7+ avec un bilan d’extension négatif et un bilan préopératoire normal. Ce patient, qu’on a vu tout à l’heure, qui avait un nodule périphérique, est éligible à un traitement radical par la chirurgie. Et donc on va lui proposer une lobectomie et un curage médiastinal.Voici la lobectomie avec le cancer qui a rétracté ici la plèvre. Le cancer à l’état fixé, en plus vu qu’on est juste à côté du bloc opératoire, on a la pièce fraiche, on a la pièce à l’état fixé.
Quand on reçoit une pièce pulmonaire, on insuffle, comme avec un tuyau d’arrosage. On insuffle le fixateur dans les bronches, pour pouvoir assurer la fixation du poumon. Car autrement par immersion simple ça ne serait pas suffisamment bien fixé. Donc on insuffle le fixateur à l’état frais dans le poumon et ensuite on fait les coupes.Ici, on a au milieu un adénocarcinome invasif et en périphérie une composante, qui est une composante in situ, qui respecte la paroi des alvéoles. On a donc un cancer mixte qui est à la fois invasif et avec une composante lipidique in situ.C’est donc un cancer que l’on va appeler invasif à prédominance lipidique.Le diagnostic histologique va aussi donner le terme de ce cancer mais aussi la classification TNM et PTNM en fonction du nombre de ganglion, de la présence de métastases et des limites de réjections par rapport à la plèvre.->Ce cancer, classé selon la TNM 8, sera donc un T2 N1 (parce qu’il y aura un ganglion envahi) et donc on total un adénocarcinome primitif pulmonaire infiltrant à prédominance lipidique PT2 a N1 R0.
On voit un aspect en dentelle normal, un aspect en dentelle plus dense et un foyer au centre très épaissi. On a plusieurs foyers tumoraux différents. Cette tumeur était accolée à la plèvre.On a un adénocarcinome donc assez dense.

Sur la macroscopie à l’état frais, on voit la capsule autour, on devine un peu les alvéoles pulmonaires. Onfait assez vite la macroscopie à l’état frais pour disséquer la lésion. Ensuite on a l’état fixé et en général l’anapath va re disséquer avant d’inclure pour pas qu’on ait beaucoup de cassettes.
On fait ensuite l'histologie, on voit au centre que c'est plus ou moins dense par rapport au reste.
On voit la tumeur qui est au niveau de la plèvre viscérale. Il y a deux composantes cettetumeur : une composante qui a un aspect plutôt infiltré dans le chorion et plus loin on a une partie qui est au niveau de la partie périphérique de la tumeur et on a un aspect de prolifération qui longe les alvéoles pulmonaires. C’est ce qu’on appelle un carcinome lépidique (anciennement appelé carcinome broncholo- alvéolaire), c’est un aspect cubo-prismatique (pas des pavimenteuses) mais elles poussent le long des alvéoles. Elle remplace le tissus
normal alvéolaire par de la tumeur. Donc on a une composante comme ça in situ qui longe les alvéoles et on a au centre de la tumeur, des tumeurs qui infiltrent vraiment le chorion.
Chez ce patient qui a été opéré on va rien faire en test moléculaire, parce que on avait un cancer localement peu avancé, qui a bénéficié d’une exérèse et si il est dans le bon groupe la survie à 5ans peut être de 90%. Malheureusement, ce type de patient est relativement rare dans le cancer du poumon, on a plutôt des patients avec des maladies avancées.Il y a des facteurs pronostics des cancers, cad des types histologiques qui influent le grade de différenciation, les emboles, mais il y a surtout des facteurs prédictifs.
• Les carcinomes à petites cellules, non à petites cellules, ne nécessitent pas de polychimiothérapie de type CPC.
• Les cancers non épidermoïdes sont traités par des sels de platines• Les adénocarcinomes sont traités par des thérapies ciblées.
Les thérapies ciblées sont effectivement selon un cas, le fait de déterminer ces cibles moléculaires, mutations cardiaques, egfr, R2 etc et on trouve 12% de mutations activatrices de l’egfr dans les

adénocarcinomes pulmonaires. Cela permet donc d’envisager une thérapie ciblée

Conclusion anatomopathologique :
Adénocarcinome primitif pulmonaire infiltrant, à prédominance lépidique (OMS 2015) Classification pT2aN1R0 (TNM 2017)
Cad que les marges de restrictions sont saines. Donc on a un carcinome bien différencié. On va avoir une prise en compte qui va prendre en compte le type histologique, le grade de différenciation, le fait que ça envahisse ou pas des ganglions, des vaisseaux, etc.
Elle est N1 car on a trouvé un envahissement au niveau des ganglions type intra pulmonaire et hilaire.
On va avoir aussi un rôle du pathologisme cellulaire, selon le stade tumoral, on va aller chercher des lésions moléculaires qui vont prédire ou pas la réponse à des thérapies ciblées.
Qu’est-ce que sont c’est thérapies ciblées ? Quand et pourquoi les utilise-t-on ?
On utilise des thérapies ciblées :- soit dans le cancer du sein qui est herceptin positif qui va d’emblé être traiter par du stratuzubam- Soit le plus souvent pour des malades qui ont un cancer avancé ou métastasique
Dans le cancer du sein, quand on a un cancer basale ou R2 positif, si on avait l’arme atomique, on l’utiliserait car on sait qu’il ne faut pas attendre. On n’attend pas le stade métastasique, on sait que les femmes doivent être traitées par le traitement le plus lourd possible. C’est un peu comme dans les leucémies où on cherche à éradiquer d’emblé les cellules cancéreuses.

Dans le cancer colorectal ou dans le cancer du poumon, on attend le stade métastasique pour dégainer les analyses génétiques et les thérapies ciblée. C’est pas pareil pour le cancer du sein R2Positif ou de type basale, où on va d’emblé, même à un stade peu avancé, faire un traitement important (chimiothérapie, hormonaux thérapie adjuvante, R2 si possible etc).Pour les cancers colorectaux et du poumon on va attendre le stade métastatique ou avancé pour dégainer un traitement général.
C’est traitements général sont de deux ordres :• Soit des anticorps anti egfr extracellulaire• Soit des inhibiteurs de tyrosine kinase qu’on appelle des nib
Dans les adénocarcinomes pulmonaires métastasiques, les nib, les erlotinib ou les gefitinib, vont inhiber la croissance des cellules tumorales cancéreuses quand elles présentent une mutation activatrice de l’egfr. A l’inverse les patients qui n’ont pas la mutation d’egfr ne répondent pas à ces traitements.Il est important de donner l’inhibiteur chez les patients muté pour egfr et à l’inverse chez les patients qui ne sont muté pour l’egfr de leur donner plutôt le traitement à base de sel de platine, qui est le traitement de référence. Cela nous permet de sélectionner les patients.
Place du pathologiste et du généticien/biologiste moléculaire dans la prise en charge des cancers bronchopulmonaires.
Comment se fait ce type d’analyse ?
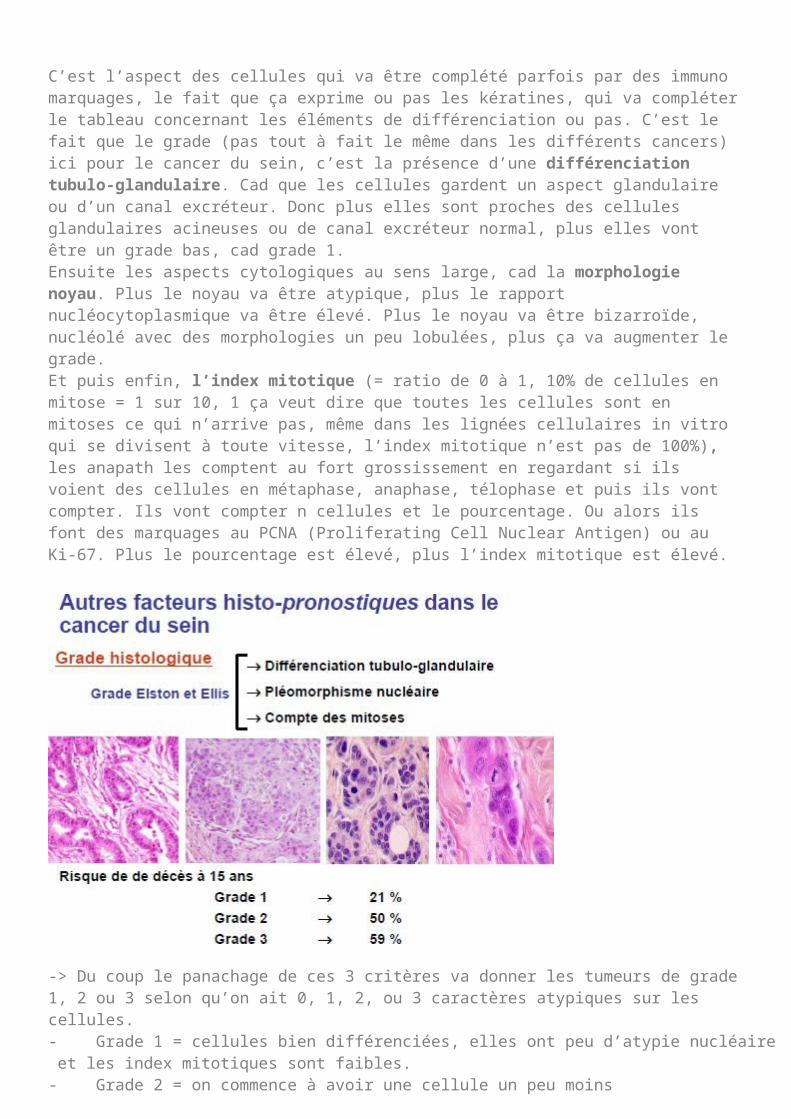
Ce type d’analyse par du clinicien qui dit au pathologiste « j’ai un patient qui a un cancer de stade avancé métastasique du cancer du poumon du colon », envoyer le bloc de la tumeur au labo de la plateforme de génétique moléculaire des cancers d’Aquitaine, dont Merlio à l’honneur d’être le coordonnateur, avec d’un côté son labo au chu de bordeaux et celui de Isabelle Suberant à l’institut Bergonié.Le service de Merlio fait les recherches des mutations à partir des bloc en paraffine et ils renvoient les résultats au pathologiste et au clinicien, que celui-ci habitent Bayonne, Périgueux, Nouméa, Pointa Pitre, Saint Denis ou Saint Pierre à la Réunion.En pratique on fait ce type d’analyse pour rechercher les mutations qui vont nous permettre la prescription de l’erlotinib, du gefitinb, d’un antialk, d’un antiroche, qui sont des marqueurs cible thérapeutique de la tumeur.
Les traitements sont différents selon le type histologique :
- Pour les carcinomes épidermoïdes (= sur base de métaplasie, qui ont remplacé un épithélium par un épithélium malpighien qui s’est transformé ensuite kératinisé ou pas) c’est compliqué car il n’existe pas beaucoup de biomarqueurs. Il y a l’amplification du Rc tyrosine kinase et EGFR1 qui pourraient préfigurer des thérapies ciblées. Mais du coup il n’existe pas trop de stratégies thérapeutiques.
- Pour les non épidermoïdes, et en particulier les adénocarcinomes, on a environ dans 10-15% des cas, des mutations de l’EGFR, du Rc à l’EGF. Du coup on a des mutations ponctuelles dans le domaine tyrosine kinase, certaines sont sensibles ou pas à des thérapies ciblées, à des inhibiteurs de tyrosine kinase. On va rechercher ces mutations. Il y a des biomarqueurs qui prédisent la sensibilité aux platines, c’est l’expression d’un certain nombre de molécules de réparation de l’ADN qui vont rendre les cellules plus ou moins sensibles. C’est très discuté, mais globalement, les cellules répondent mieux quand elles n’exprimaient pas la molécule de réparation car les cellules tumorales ont accumulées plus de lésion et donc elles mourraient davantage car elles n’étaient pas capables de réparer les lésions créées par ces médicaments.
TTF1 est un facteur de bon pronostic, puisque se sont des cellules bien différenciées en général.
On recherche les mutations dans la tumeur in sillico (sur ordinateur) et l’on fait alors un ciblogramme ou tumorogramme au niveau moléculaire.

Ici ce n’est plus des Ac bloquants qu’on va donner mais des inhibiteurs de tyrosine kinase qui vont agir soit sur EGFR soit sur PI3kinase, soit sur BRaf soit sur Alc, etc.
Il faut faire attention quand même avec les anti-EGFR car elles peuvent causer des problèmes du aux effets secondaires (au niveau épiderme,…). Cependant, ils sont moindres que ceux de la chimiothérapie.
Il a passé vite car il manquait de temps mais l’essentiel à retenir est avant !
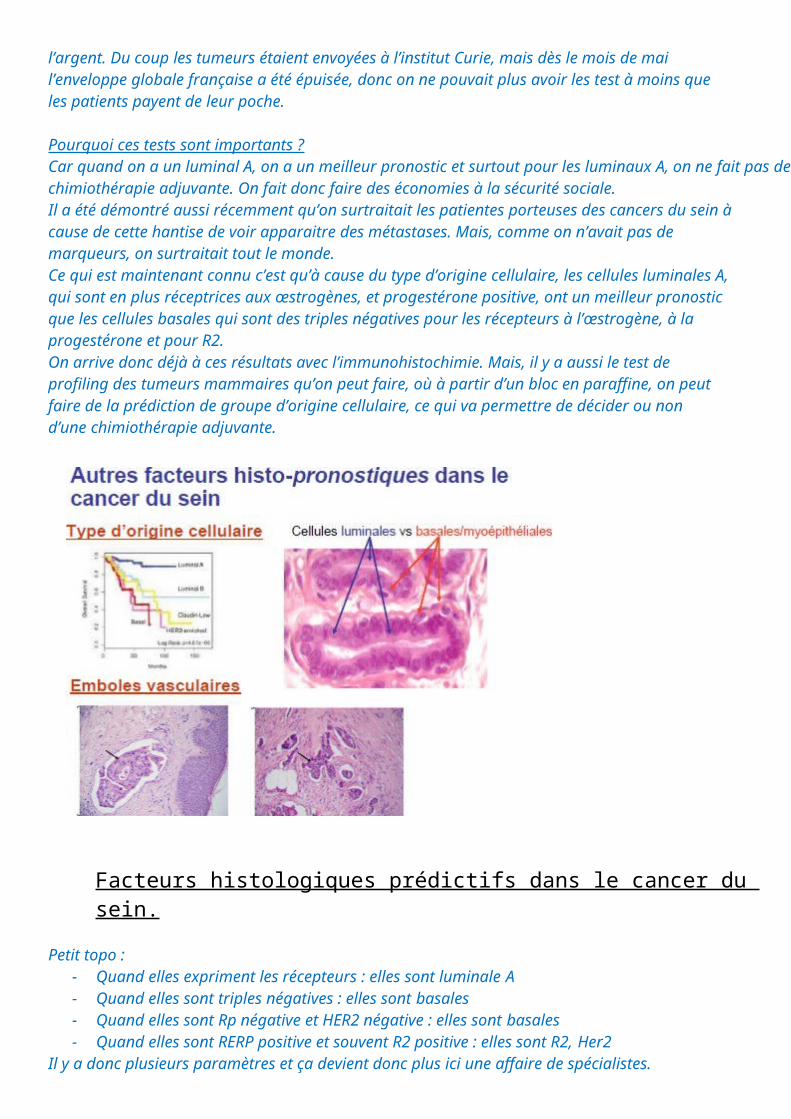
Question élève : pourquoi on n’utilise pas ça d’emblé pour tout le monde ?Car il y a d’un côté des patients qui sont opérables d’emblé, ici à la Réunion comme il n’y a pas beaucoup de patients opérables d’emblé, on envoie tous les blocs de stade avancé au labo de Merlio.Et donc on n’utilise pas ça parce que la mutation n’est observée que chez 15% des patients, donc on a un médicament pour egfr, un médicament pour Braf, un médicament pour Met et un médicament pour alk.Donc il faut identifier les patients en stade métastatiques, qui ont la mutation qui va répondre. Mais comme on peut le voir, il reste au moins 20 à 40%, selon les cas, de patients qui n’ont pas de mutation éligible pour un traitement.Et dans le domaine des carcinomes épidermoïdes, on n’a pas pour l’instant de traitement ciblé.Il y a pour terminer un dernier traitement. Il y a maintenant une nouvelle thérapeutique qui ne cible pas directement des mutations mais qui sont des thérapeutiques qui sont des immunothérapies, qui permettent au système immunitaire de venir détruire ces cellules tumorales.
- Soit il y a des traitements ciblé vers une mutation- soit on va utiliser ce qu’on appelle des immunothérapeutiques (ça c’est toujours pour les patientes
métastasiques. Les patients qui sont opérables, on va d’abord faire l’opération)- Les patients qui n’ont aucun marqueur de réponse on va leur faire une chimiothérapie à base de sel
de platine.





























