Embed Size (px)
Citation preview

Erythroblastopénies
constitutionnelles & acquises
Thierry LEBLANC
Hôpital Robert-Debré & Saint-Louis
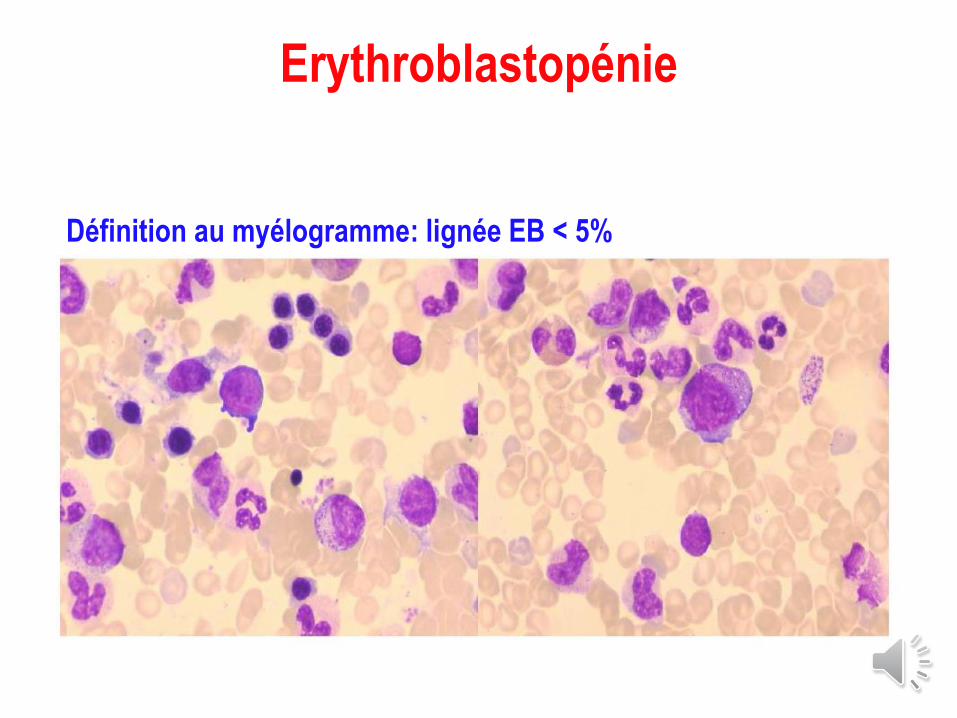
Erythroblastopénie
Définition au myélogramme: lignée EB < 5%

Etiologie des érythroblastopénies
Et autres…(dont maladies métaboliques:
acidurie méthylmalonique)
(Means T, ASH 2016)
+ Alloimmunes?

Erythroblastopénie transitoire de l’enfant
Anémie sévère de la petite enfance (< 4 ans)
Neutropénie associée fréquente + thrombocytose
Diagnostic souvent fait au nadir de l’Hb : réticulocytose peut être présente +++
mais: pas de macrocytose, d’élévation HbF et de l’ADAe (+ pas d’ATCD Fam, de malformations,…)
Besoins transfusionnels parfois
Origine virale mais virus en cause ?
Guérison : délai < 2 mois post diagnostic

N = 36 : étude monocentrique sur 30 ans
Hémoglobine médiane au diagnostic: 4,4 g/dL
Patients transfusés: 72%
? +++ variation épidémiologique ? :
(Acta Paeditrica, 2014)

Crise aplastique d’une AH
Mode de révélation non rare d’une AH
constitutionnelle : SH +++ ou non
(AHAI)
Virus en cause : parvovirus B19
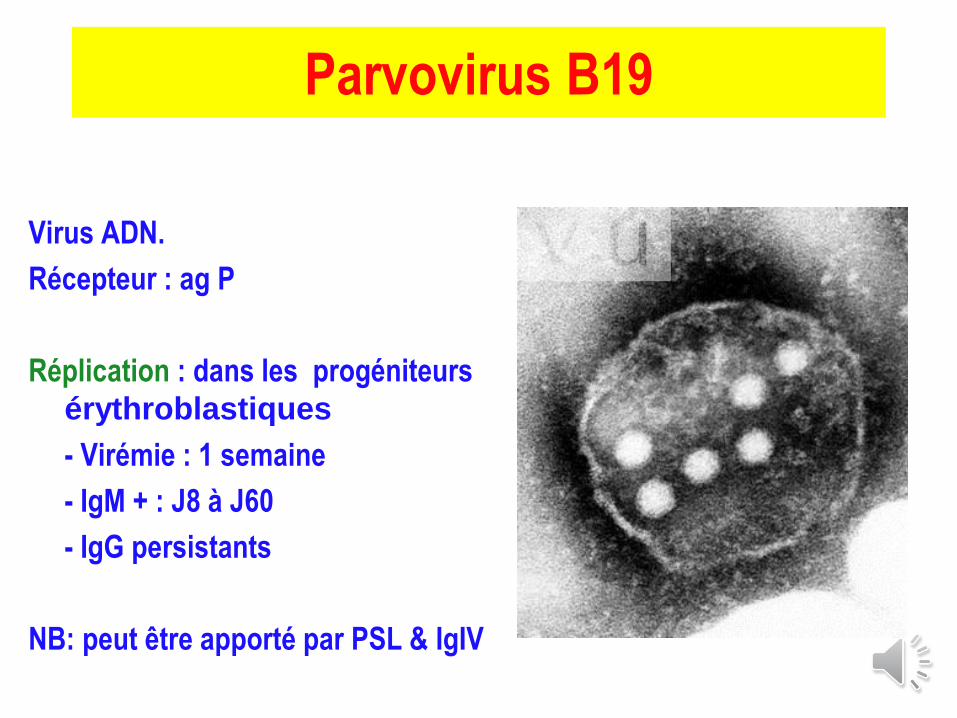
Parvovirus B19
Virus ADN.
Récepteur : ag P
Réplication : dans les progéniteurs
érythroblastiques
- Virémie : 1 semaine
- IgM + : J8 à J60
- IgG persistants
NB: peut être apporté par PSL & IgIV

Parvovirus B19 et anémie (1)
Non anémiant chez le volontaire sain après inoculation
Arrêt de l’érythropoïèse en 7-10j
Reprise rapide
Au total : baisse Hb limitée: 1 à 2 g/dL
Anémie subaiguë chez les pts avec AH
Atteinte hématologique plus globale possible chez des pts IS

Parvovirus et anémie (2)
Médecine fœtale et NN
Anasarque fœtoplacentaire(cycle lytique au niveau des précurseurs érythroïdes du foie
fœtal)
Anémie néonatale par infection chronique
Diagnostic différentiel avec l’ABD
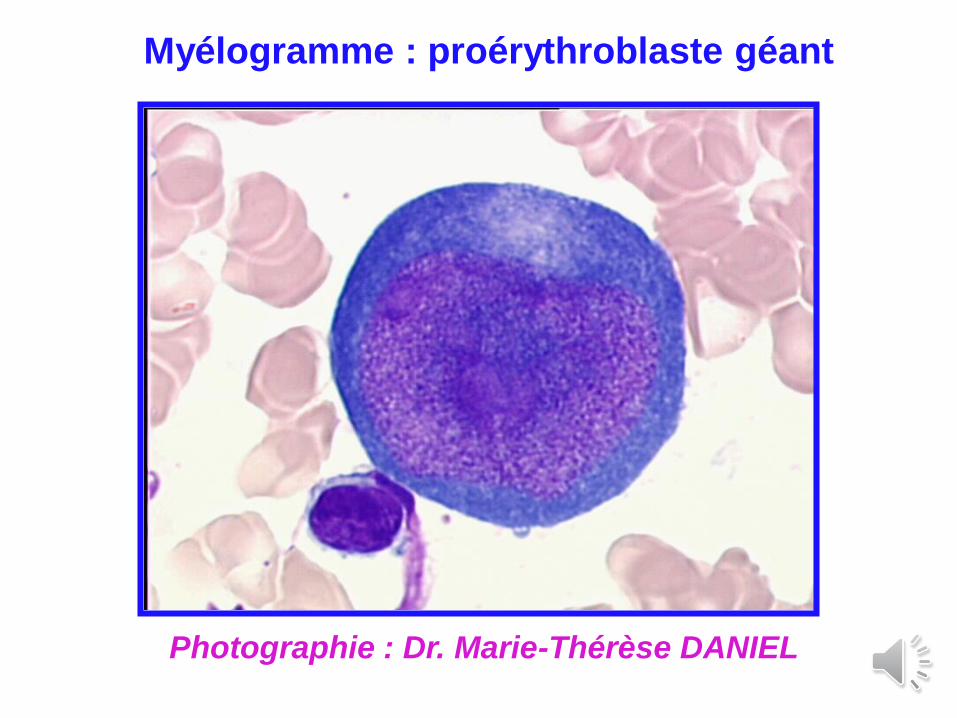
Myélogramme : proérythroblaste géant
Photographie : Dr. Marie-Thérèse DANIEL

Infection chronique par le PVB19
Fœtus et Nné
Immunosuppression au sens large:
- primitives: DI
- Secondaires: traitements IS, chimiothérapie, allogreffe,... & VIH
Diagnostic : - myélogramme : proérythroblastes géants
- tests virologiques : PCR sur la moelle
Traitement : IgIV

Erythroblastopénies auto-immunes
(très rares +++ en pédiatrie)
Contexte ± franc d’IS (ou d’hémopathie) chez
l’adulte
Peut apparaître isolée et être inaugurale ou
révélatrice d’une atteinte sous-jacente.
Si PRCA isolée: bilan étiologique poussé +++

Diagnostic d’une
érythroblastopénie autoimmune
Valeur :
- d’un contexte autoimmun connu ou identifié: AHAI, LED, myasthénie, …
NB : rechercher un thymome
- d’une prise de médicament: ex: Epo
- des cultures médullaires : • pousse Nle in vitro
• éventuellement: inhibition pousse par le serum du pt
Traitement : immunosuppresseurs

Erythroblastopénie & AHAI
Pas exceptionnelle (LED & autres)
Diagnostic évoqué sur la réticulocytopénie et
confirmé par le myélogramme
Sévérité +++ de l’anémie initialement: support
en CG prolongé: temps d’action du
traitement IS & sortie des réticulocytes

Erythroblastopénie & thymome
Thymomes: < 5% associés à une érythroblastopénie
Erythroblastopénie: 7 à 10% sont dues à un thymome
Diagnostic: TDM
Traitement:
- Classiquement: thymectomie mais:• RC pas constante
• Rechute possible après exérèse
- Immunosuppression
Thymectomie rediscutée

Erythroblastopénie & hémopathies lymphoïdes
LLC, LGL, Waldenstrom, LNH & Hodgkin,…
Physiopathologie le plus souvent AI (mais pas
toujours B)
Bilan initial orienté +++: frottis, immunophénotype
sang, recherche de clone B ou T,…
Bénéfice au traitement de la maladie de fond +++

Erythroblastopénie & médicaments
Liste non exaustive
Physiopathologie pas toujours
connue:
- Hématotoxicité directe: exemple:
thiamphenicol
- Autoimmune (AC)
Un modèle très bien étudié +++:
érythroblatopénie post Epo
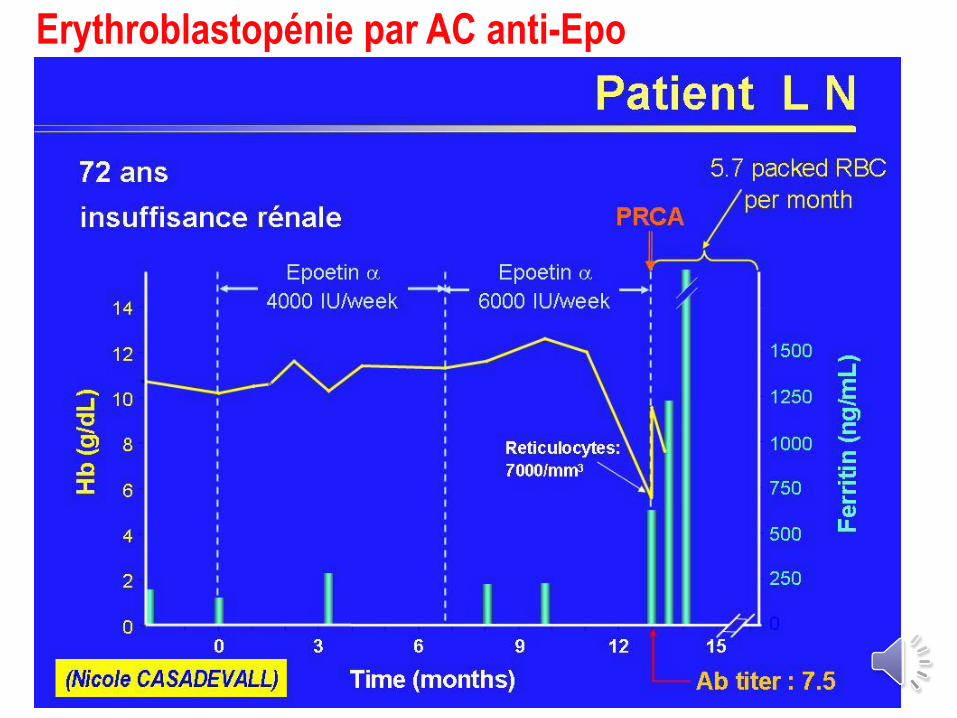
Erythroblastopénie par AC anti-Epo
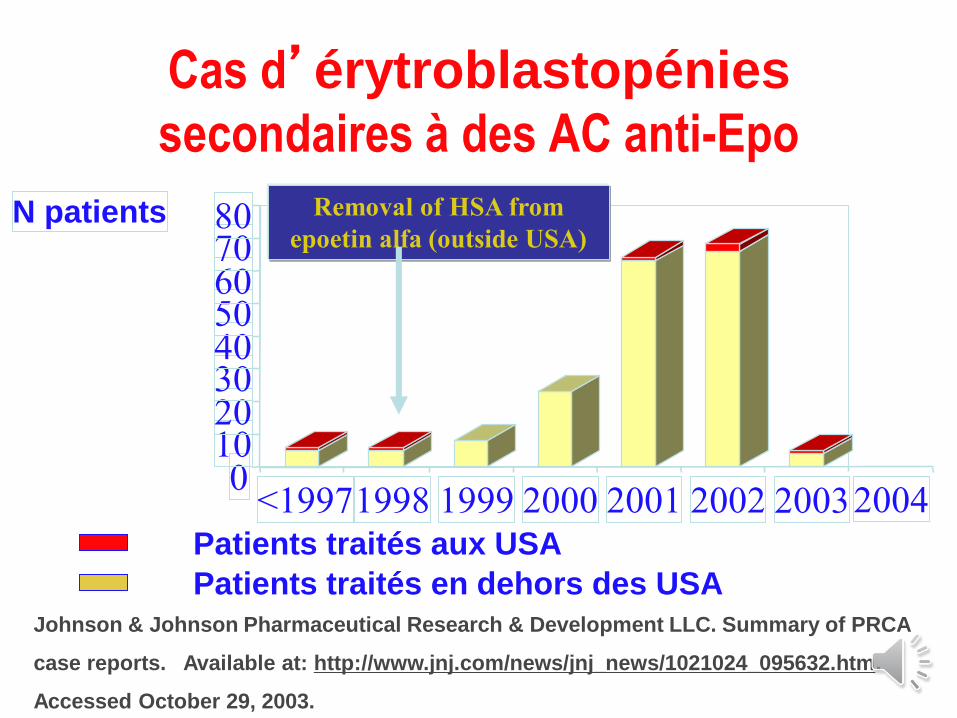
Cas d’érytroblastopénies
secondaires à des AC anti-Epo
01020304050607080N patients
<19971998 1999 2000 2001 2002 2003
Removal of HSA from
epoetin alfa (outside USA)
2004Patients traités aux USA
Patients traités en dehors des USAJohnson & Johnson Pharmaceutical Research & Development LLC. Summary of PRCA
case reports. Available at: http://www.jnj.com/news/jnj_news/1021024_095632.htm.
Accessed October 29, 2003.

Erythroblastopénie par AC a Epo
Cas tous rapportés chez des pts avec IR
(2 cas de SMD)
Corrélation +++ avec l’EPREX® donnée par voie
SC (pas de cas si voie IV exclusive)
Délai médian : 11 mois (3-90)
AC neutralisants dirigés contre le core
AC inhibant les cultures érythroïdes
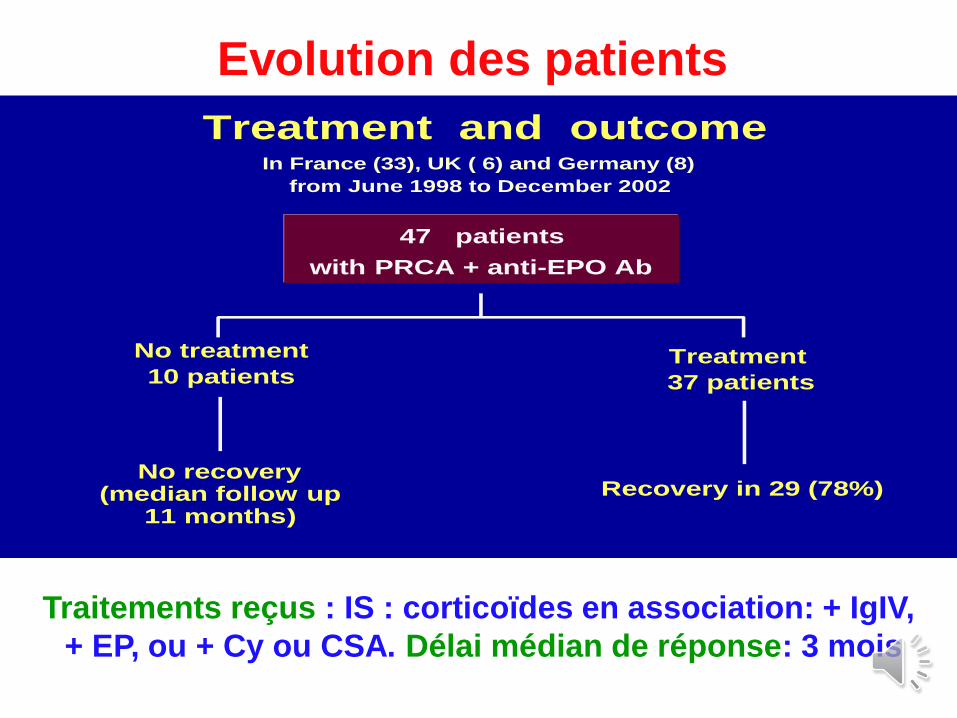
Treatment and outcomeIn France (33), UK ( 6) and Germany (8)
from June 1998 to December 2002
47 patients
with PRCA + anti-EPO Ab
No treatment
10 patientsTreatment
37 patients
No recovery(median follow up
11 months)
Recovery in 29 (78%)
Evolution des patients
Traitements reçus : IS : corticoïdes en association: + IgIV,
+ EP, ou + Cy ou CSA. Délai médian de réponse: 3 mois
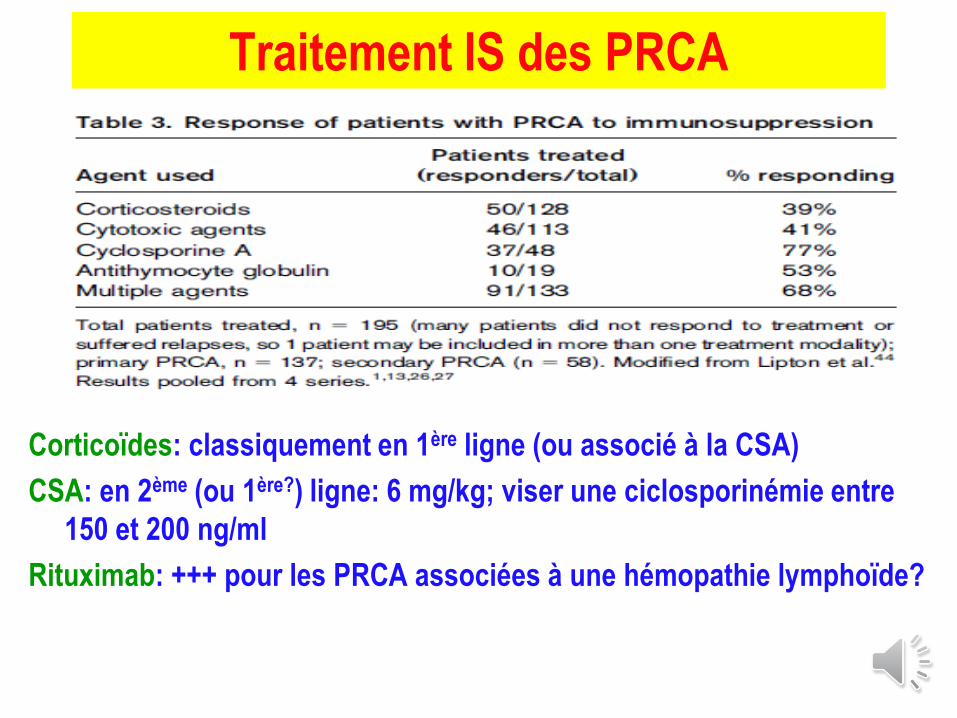
Traitement IS des PRCA
Corticoïdes: classiquement en 1ère ligne (ou associé à la CSA)
CSA: en 2ème (ou 1ère?) ligne: 6 mg/kg; viser une ciclosporinémie entre
150 et 200 ng/ml
Rituximab: +++ pour les PRCA associées à une hémopathie lymphoïde?

Anémie de Blackfan-Diamond

Autres érythroblastopénies
constitutionnelles?
1. Problématique GATA1
2. Mutation du gène de l’EPO: 1 famille
(consanguinité; 2 pts d’une même fratrie).
R150Q homozygote: Glu Arg (Ah Ram Kim, Cell 2017)
3. Mutations de CECR1 (syndrome DADA2)
4. Mutations de gènes favorisant l’autoimmunité
Ex: 1 cas de PRCA chez une adolescente mutée pour CTLA4
(Dybedal & al, Blood 2015)

CECR1 & erythroblastopénieCECR1: code pour l’ADA2(impliqué dans le syndrome DADA2: atteinte rhumatismale & déficit immunitaire)
Allemagne: 68 pts (-): 4 mutations bi-alléliques de CECR1
France: 2 pts identifiés à ce jour
Turquie +++
Profil particulier:
Anémie: non régénérative avec besoins transfusionnels: âge médian au diagnostic: 5 semaines [0-14 ans].
NB: HbF normale
Extra-hématologique: malformations possibles, hypogammaglobulinémie +++, polysomes normaux, CT peu actifs et a priori CI, pas de réponse aux inhibiteurs du TNFa
indication +++ d’allogreffe
(ASH 2017, Wlodarski & al)

T. Leblanc 2018
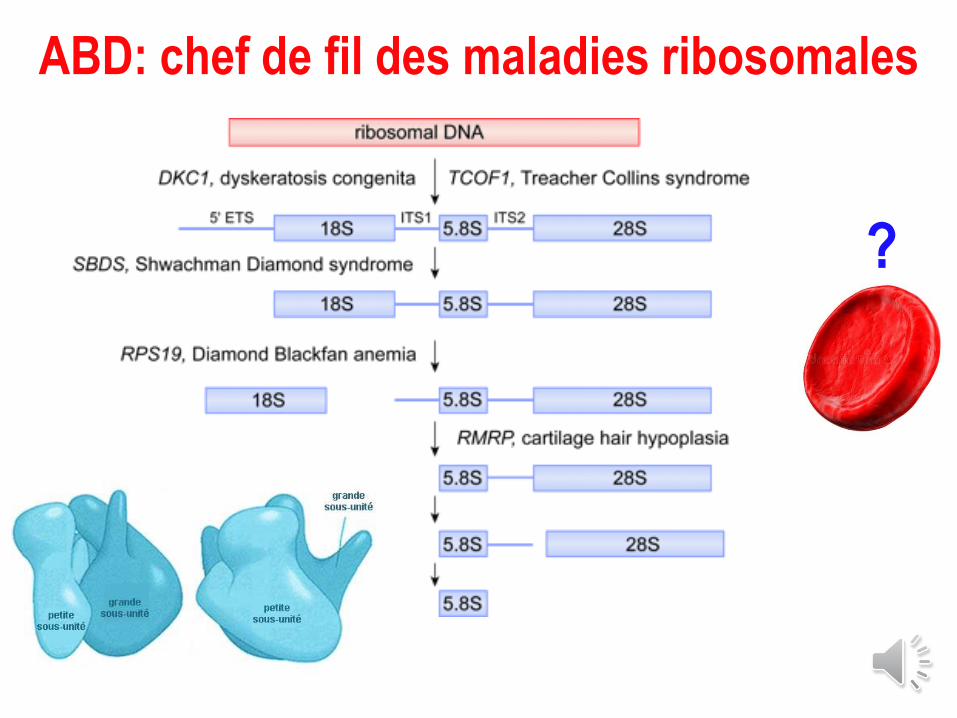
ABD : mots clés
Maladie génétique rare
Transmission classiquement AD mais… pas si simpleNB: 2 exceptions GATA1 et TSR2 : transmission liée à l’X
Première cause d’érythroblastopénie de cause génétique
Grande hétérogénéité clinique et génétique
►Conseil génétique complexe

Diagnostic différentiel
►Autres érythroblastopénies
- Erytroblastopénie transitoire (TEC)
- Crise aplastique d’une AH
- Infection chronique au PVB19
- Autres érythroblastopénies acquises
►Autres anémies arégénératives (GATA1, ASC,…)
►Autres IBMF: AF +++ ; pb: ABD souvent non évoquée devant un sy. polymalformatif…
(Dc d’une anémie NN ou d’âge < 2 ans)

T. Leblanc 2018
ABD : épidémiologie
Incidence (registres européens) :
- 4 à 7 cas / 1.000.000 naissances
Majorité des cas (> 75%) : sporadiques(si analyse sur le phénotype clinique/hématologique)
Cas familiaux
France en 2016:785.000
(données INSEE)

ABD : génétique (1)
Maladie autosomique dominante mais:
rares cas liés au sexe: mutations de GATA-1 & de TSR2 (gènes portés par le chr X)
pénétrance variable: phénotype silencieux (porteur sain de la mutation)
phénomène d’anticipation génétique?
De plus en plus de gènes…: 24 gènes RP (RPS & RPL) + TSR2 + GATA1
• 1er gène: RPS19: 46,XX, t(X;19)(p21;q13) : DBA1: muté chez 25% des pts
• Depuis très nombreux autres gènes identifiés codant pour des protéines de la petite ou de la grande sous-unité + GATA-1 & TSR2
• MAIS il reste une proportion non nulle de pts sans mutation/délétion identifiée
Quelques corrélations génotype / phénotype
Conseil génétique difficile +++ donc « maximaliste »: risque de 50% et gravité imprévisible (malformations, hématologie)

Diagnostic génétique: Pr. Lydie DA COSTA
Laboratoire d’hématologie, CHU Robert-Debré, Paris.
Etape 1: recherche de variations alléliques par NGS(Librairie Roche “NimbleGen SeqCap EZ” et puce illumina (Flowcell
standard 2*150) et passage sur un Miseq/Nextseq)
Etape 2: recherche de grandes délations (15 à 20% des pts) par CGH/SNP
(HumanOmniExpress-12 v1.0 Analysis BeadChip Kit (>700 000 loci) / Genome studio software. Puces Custom)
Etape 3: séquençage d’exomes (triplets)(plateforme Imagine, Necker-Enfants-malades)
ABD : génétique (2)
Savoir être patient… mais:
1) Dc non génétique
2) PEC idem (sauf GMO &
DPN/DPI)
ABD: chef de fil des maladies ribosomales
?
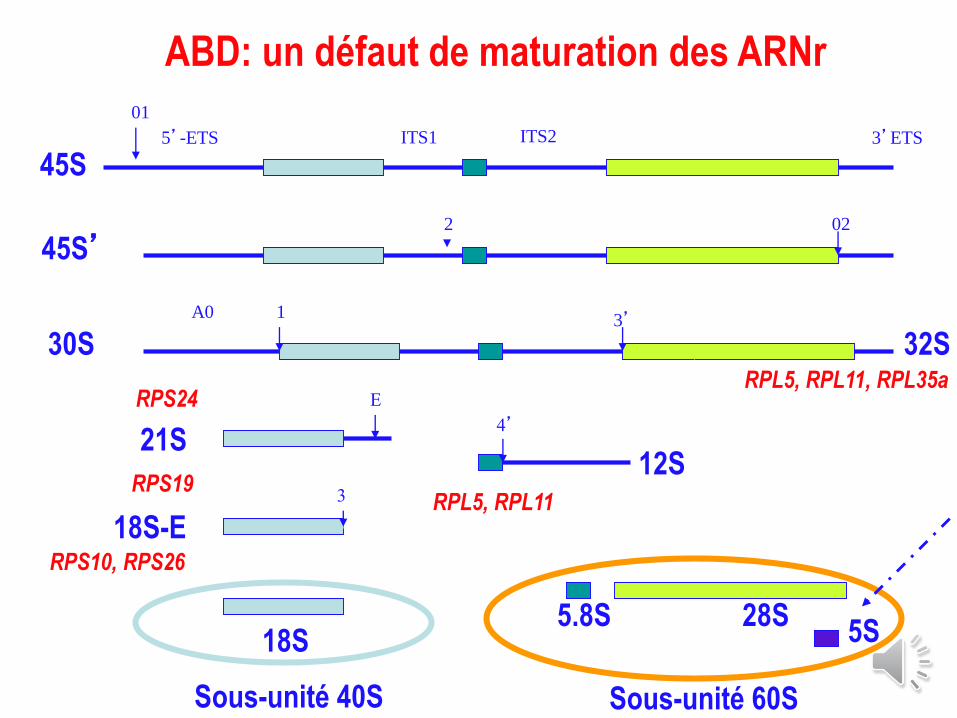
45S5’-ETS ITS1 ITS2 3’ETS
01
45S’2 02
30SA0 1 3’
32S
RPS24
21S
18S-E
RPS19
E
3
18S
Sous-unité 40S
RPL5, RPL11, RPL35a
4’
12S
5.8S 28S
Sous-unité 60S
RPL5, RPL11
RPS10, RPS26
5S
ABD: un défaut de maturation des ARNr
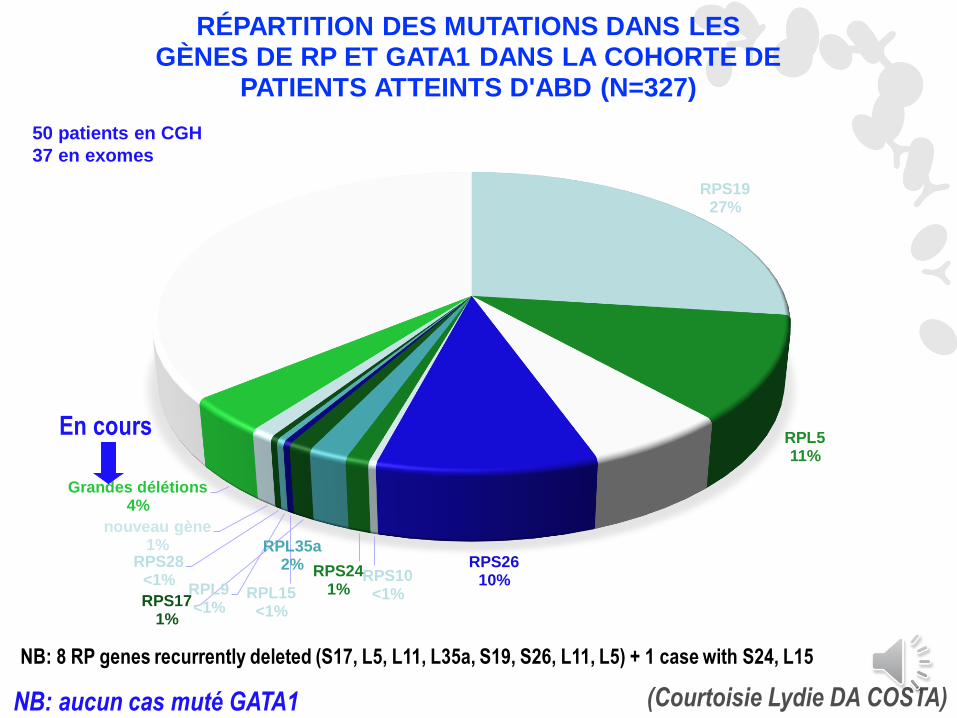
50 patients en CGH
37 en exomes
RPS1927%
RPL511%
RPL116%
RPS2610%RPS10
<1%
RPS241%
RPL35a2%
RPS171%
RPL15<1%
RPL9<1%
RPS28<1%
nouveau gène1%
Grandes délétions 4%
sans génotype36%
RÉPARTITION DES MUTATIONS DANS LES GÈNES DE RP ET GATA1 DANS LA COHORTE DE
PATIENTS ATTEINTS D'ABD (N=327)
(Courtoisie Lydie DA COSTA)NB: aucun cas muté GATA1
En cours
NB: 8 RP genes recurrently deleted (S17, L5, L11, L35a, S19, S26, L11, L5) + 1 case with S24, L15

ABD génétique: résumé
70% patients : mutation identifiée:
- 20 gènes RP (+++ RPS19); transmission AD
- 2 gènes non RP mais associés à la biosynthèse des ribosomes:
GATA1 & TSR2; transmission liée à l’X
- 2 gènes impliqués dans des phénocopies de l’ABD: CECR1 & EPO; transmission autosomique récessive
NB: 96% des mutations: 6 genes: RPS19, RPL5, RPLL11, RPS26, RPL35A, RPSS17
30% patients sans mutation: approche de type exome
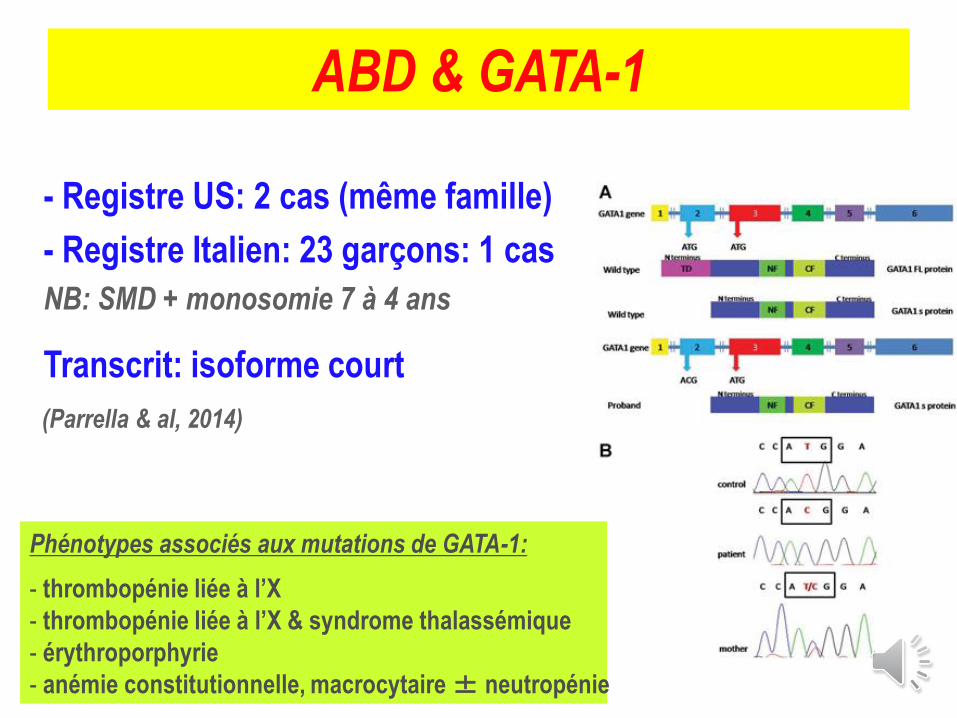
ABD & GATA-1
- Registre US: 2 cas (même famille)
- Registre Italien: 23 garçons: 1 cas
NB: SMD + monosomie 7 à 4 ans
Transcrit: isoforme court
Phénotypes associés aux mutations de GATA-1:
- thrombopénie liée à l’X
- thrombopénie liée à l’X & syndrome thalassémique
- érythroporphyrie
- anémie constitutionnelle, macrocytaire ± neutropénie
(Parrella & al, 2014)
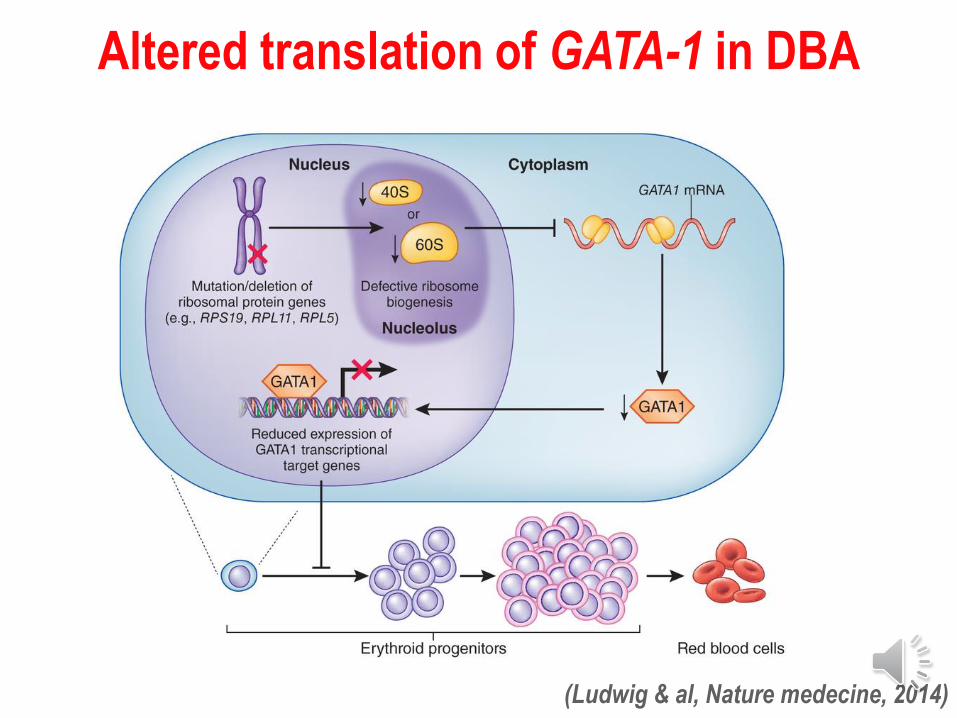
Altered translation of GATA-1 in DBA
(Ludwig & al, Nature medecine, 2014)

ABD: modalités diagnostiques
Clinique +++
NB: exclure PVB19
(Vlachos & al, BJH 2008)
(si non transfusé)
(après 6 mois)
Seul "vrai" Dc
différentiel: TCE

ABD : aspects cliniques (1)
Caractéristiques de l’anémie
Précoce : avant l’âge de 2 ans (et +++ pt NRS)
Profonde : indication transfusionnelle quasi systématique
Caractéristiques:
- macrocytaire le + souvent
- arégénérative: réticulocytes effondrés voire absents
- associée à des stigmates d’érythropöièse fœtale (+++ HbF)
- élévation de l’ADAe (si activité évaluée avant toute transfusion)
- autres lignées + normales
(neutropénie, thrombopénie ou thrombocytose possibles)
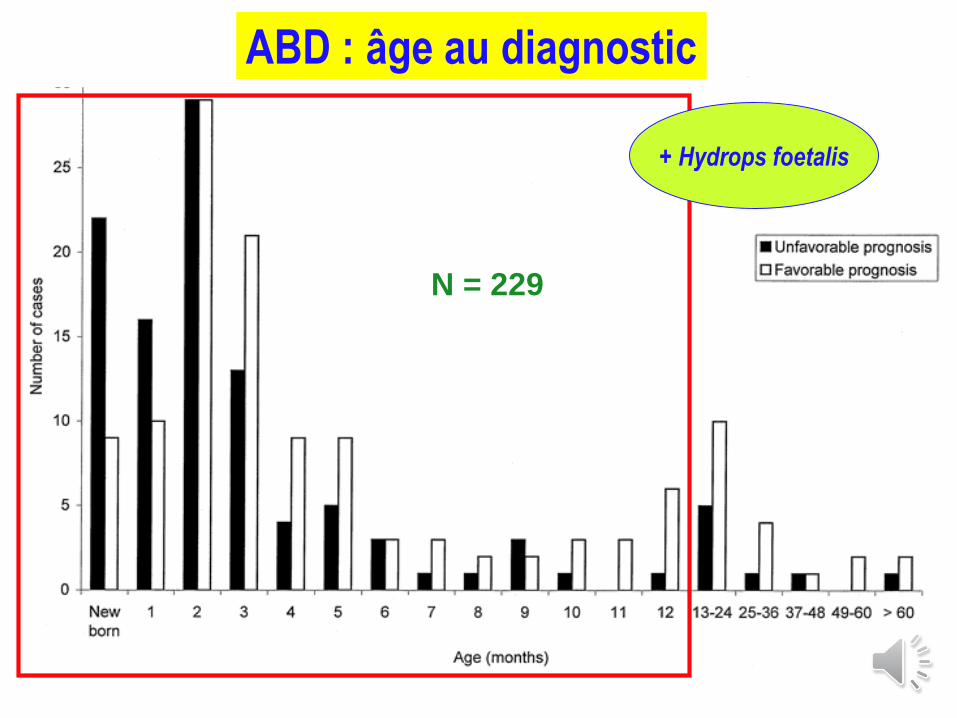
ABD : âge au diagnostic
N = 229
+ Hydrops foetalis

T. Leblanc 2018
ABD : aspects cliniques (2)
Phénotype associé très variable :
- Absent
- RCIU & RSP
- « Dysmorphie faciale »
- Eléments malformatifs : jusqu’à 40% des pts
►syndromes polymalformatifs sévères rapportés
- Tableaux: pseudo Turner, Fanconi, Treacher-Collins,…
NB : fratrie : polymorphisme clinique
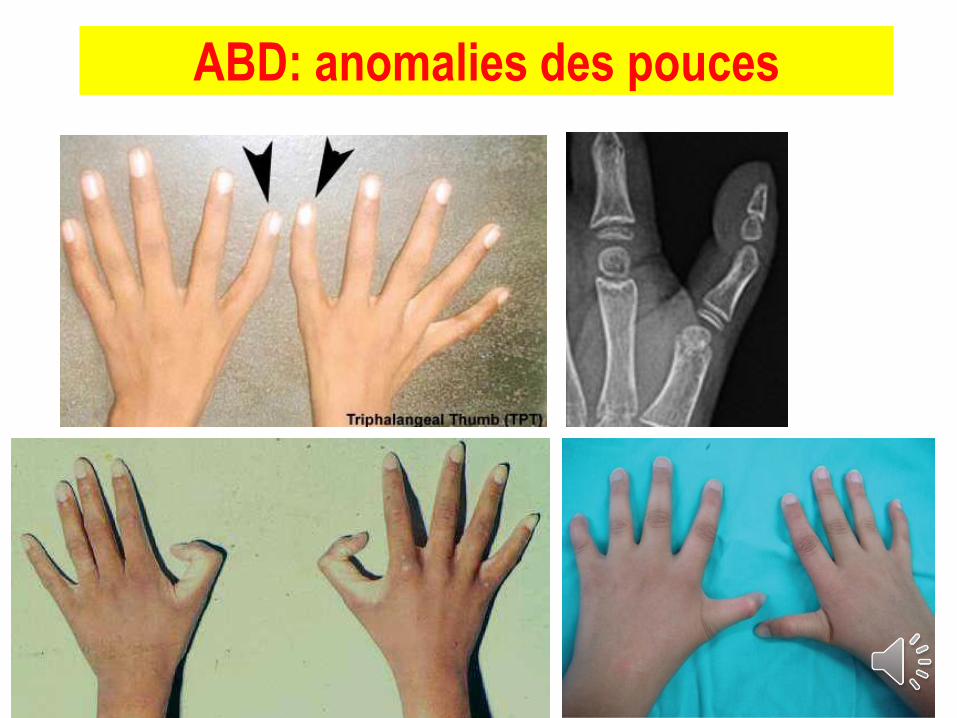
ABD: anomalies des pouces

T. Leblanc 2018
ABD: problème des phénotypes silencieux
Données génétiques: mutation sans phénotye
Données biologiques: phénotypes a minima: ADAe ,
VGM, HbF…
Données cliniques (post GMO)
?

Environ 20% des pts ont une « rémission »
Mécanisme:
- ? Le plus souvent (NRS, puberté,…
- Mosaïcisme somatique: 1 cas démontré
ABD : évolutions hématologiques (1)
(Venugopal & al, Hematologica 2017)
Mutation chez le patient: délétion 12q
(184 kb): 11 gènes dont RPS26 & RPL41
Correction partielle de l’allèle muté
(ici maternel) par l’allèle paternel
Avantage prolifératif
Expansion clonale

T. Leblanc 2018
ABD : évolutions hématologiques (2)
Complications onco-hématologiques
- leuconeutropénie / thrombopénie
- risque d’insuffisance médullaire & d’hématotoxicité
- risque augmenté de SMD/LA ( LAM)
NB: risque de perte de sensibilité aux CT avec l’âge
+
1) Mise en place d’un déficit B type DCV
2) risque augmenté de KLien biosynthèse
des ribosomes & p53
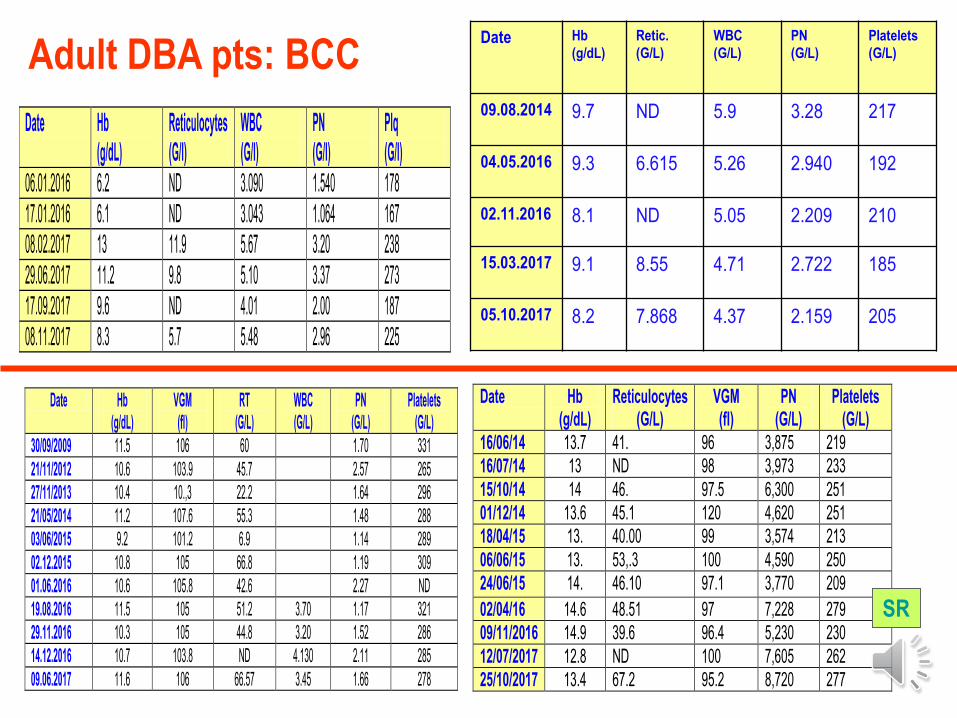
Date Hb (g/dL)
Reticulocytes (G/l)
WBC (G/l)
PN (G/l)
Plq (G/l)
06.01.2016 6.2 ND 3.090 1.540 178
17.01.2016 6.1 ND 3.043 1.064 167
08.02.2017 13 11.9 5.67 3.20 238
29.06.2017 11.2 9.8 5.10 3.37 273
17.09.2017 9.6 ND 4.01 2.00 187
08.11.2017 8.3 5.7 5.48 2.96 225
Date Hb
(g/dL) Reticulocytes
(G/L) VGM (fl)
PN (G/L)
Platelets (G/L)
16/06/14 13.7 41. 96 3,875 219
16/07/14 13 ND 98 3,973 233
15/10/14 14 46. 97.5 6,300 251
01/12/14 13.6 45.1 120 4,620 251
18/04/15 13. 40.00 99 3,574 213
06/06/15 13. 53,.3 100 4,590 250
24/06/15 14. 46.10 97.1 3,770 209
02/04/16 14.6 48.51 97 7,228 279
09/11/2016 14.9 39.6 96.4 5,230 230
12/07/2017 12.8 ND 100 7,605 262
25/10/2017 13.4 67.2 95.2 8,720 277
Date Hb
(g/dL)
Retic.
(G/L)
WBC
(G/L)
PN
(G/L)
Platelets
(G/L)
09.08.2014 9.7 ND 5.9 3.28 217
04.05.2016 9.3 6.615 5.26 2.940 192
02.11.2016 8.1 ND 5.05 2.209 210
15.03.2017 9.1 8.55 4.71 2.722 185
05.10.2017 8.2 7.868 4.37 2.159 205
Date Hb (g/dL)
VGM (fl)
RT (G/L)
WBC (G/L)
PN (G/L)
Platelets (G/L)
30/09/2009 11.5 106 60 1.70 331
21/11/2012 10.6 103.9 45.7 2.57 265
27/11/2013 10.4 10.,3 22.2 1.64 296
21/05/2014 11.2 107.6 55.3 1.48 288
03/06/2015 9.2 101.2 6.9 1.14 289
02.12.2015 10.8 105 66.8 1.19 309
01.06.2016 10.6 105.8 42.6 2.27 ND
19.08.2016 11.5 105 51.2 3.70 1.17 321
29.11.2016 10.3 105 44.8 3.20 1.52 286
14.12.2016 10.7 103.8 ND 4.130 2.11 285
09.06.2017 11.6 106 66.57 3.45 1.66 278
SR
Adult DBA pts: BCC
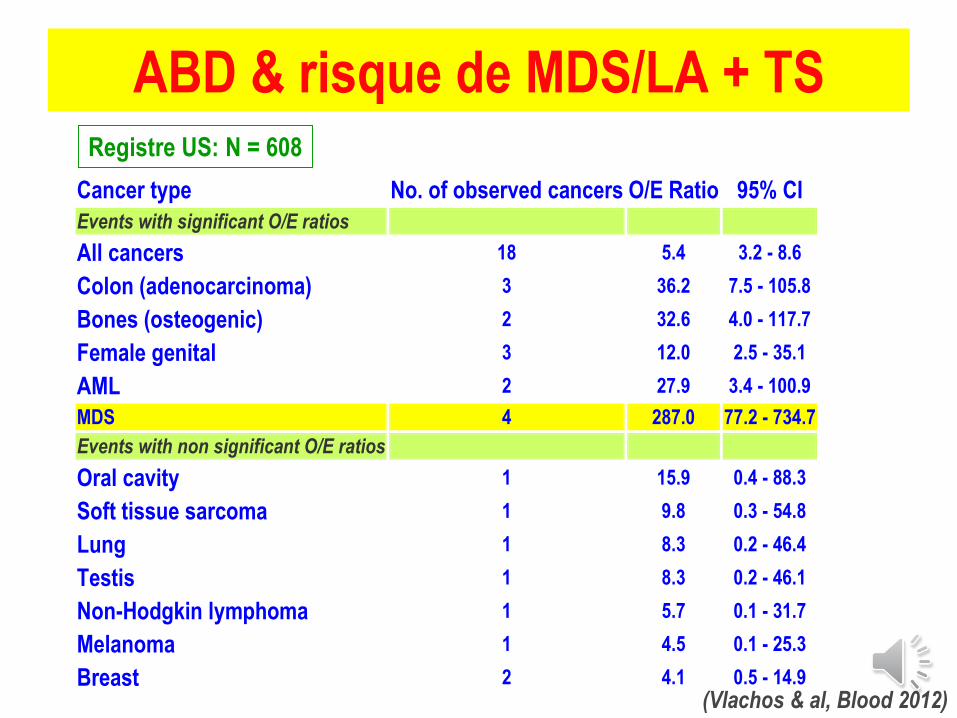
Cancer type No. of observed cancers O/E Ratio 95% CI
Events with significant O/E ratios
All cancers 18 5.4 3.2 - 8.6
Colon (adenocarcinoma) 3 36.2 7.5 - 105.8
Bones (osteogenic) 2 32.6 4.0 - 117.7
Female genital 3 12.0 2.5 - 35.1
AML 2 27.9 3.4 - 100.9
MDS 4 287.0 77.2 - 734.7
Events with non significant O/E ratios
Oral cavity 1 15.9 0.4 - 88.3
Soft tissue sarcoma 1 9.8 0.3 - 54.8
Lung 1 8.3 0.2 - 46.4
Testis 1 8.3 0.2 - 46.1
Non-Hodgkin lymphoma 1 5.7 0.1 - 31.7
Melanoma 1 4.5 0.1 - 25.3
Breast 2 4.1 0.5 - 14.9
ABD & risque de MDS/LA + TS
(Vlachos & al, Blood 2012)
Registre US: N = 608
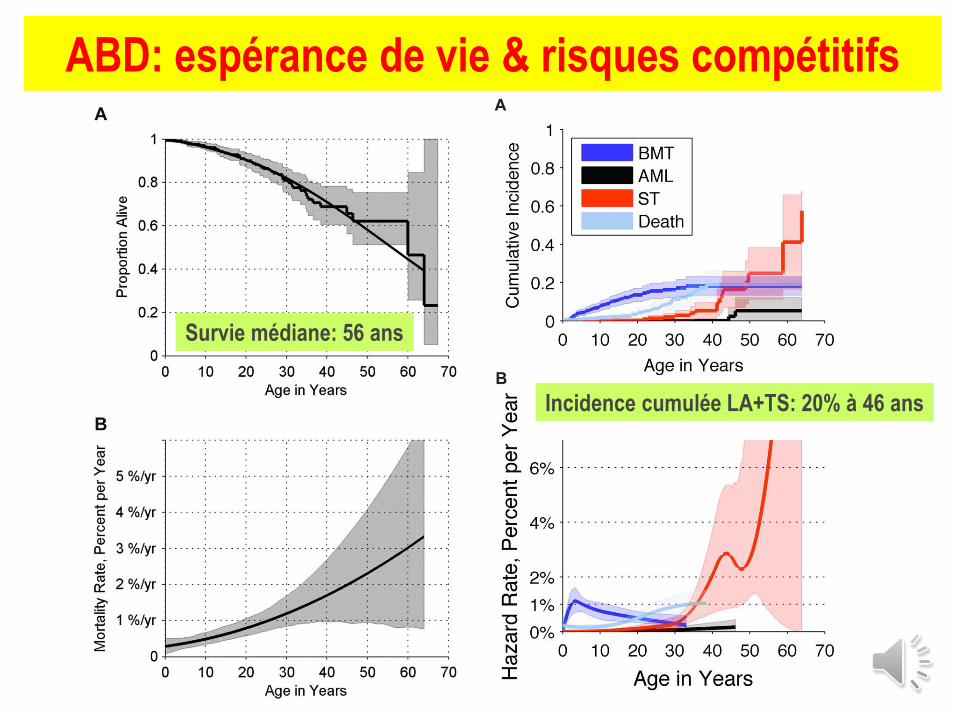
ABD: espérance de vie & risques compétitifs
Survie médiane: 56 ans
Incidence cumulée LA+TS: 20% à 46 ans

T. Leblanc 2018
ABD & risque de cancer
A confirmer: problème des cohortes adultes & des
biais d’inclusion
Pas de recommandations établies en la matière:
- risque a priori pas « majeur »
- spectre des TS très variable
Surveillance & hygiène de vie…

T. Leblanc 2018
ABD : aspects évolutifs extra- hématologiques
Problème des grossesses

T. Leblanc 2018
OFBADObservatoire Français de l’ABD
Base de donnée INTERNET finalisée avec l’unité d’épidémiologie clinique (Pr. C. Alberti, R. Debré)
Enquête épidémiologique et clinique parallèle au génotypage de la cohorte Française (PHRC); objectif: rechercher des corrélations phénotype/génotype

Anémie de Blackfan-Diamond
Prise en charge & Traitement

T. Leblanc 2013
Prise en charge d’un enfant
atteint de DBA
Faire le diagnostic:
- Clinique
- Génétique / famille
Traiter l’anémie:
- Transfusions
- Test aux corticoïdes
Inclure dans l’observatoire
Prévenir les effets
secondaires du ttt +++
Dépister évolutions H ou K
Mettre en place un suivi
pluridisciplinaire
Conseil génétique…
CRMR Aplasies médullaires & Filière MaRIH
AFMBD

Valeur des ATCD & des NFS antérieures
Myélogramme indispensable: exclure un SMD (en
sachant que l’ABD peut évoluer vers un SMD…) et
que la moelle peut être de lecture difficile…
Bilan malformatif, biologique & génétique idem.
NB: phénotype recouvrant avec les autres IBMF
Prise en charge d’un adulte suspect
d’être atteint d’ABD

T. Leblanc 2018
ABD : options thérapeutiques
• 1) Corticothérapie
• 2) Transfusions
• 3) Traitements alternatifs et expérimentaux
• 4) Greffe de CSH
• 5) thérapie génique (RPS19)

T. Leblanc 2018
ABD & corticothérapie
Traitement de référence
Posologie initiale : 2 mg/kg/j: dès l’âge d’1 an révolu
Dose max: 80 mg/kg (adulte)
Réponse réticulocytaire rapide : au 8ème/10ème jour
Corticosensibilité : 60 à 70% des pts (chez l’enfant)
Décroissance lente ensuite
Objectif : posologie < 0,5 mg/kg/j (voire < 0,25, < 0,15…)
Plateau : à passer 1j sur 2
Adulte: quelle dose maximale: 10 mg? Pas de schéma 1j sur 2 ici.

T. Leblanc 2018
ABD corticothérapie: principes
.Test systématique dès que l’enfant a > 1 an
. Traitement poursuivi si dose tolérable
. Posologie peut souvent être 2dairement diminuée
. Faut il arrêter la corticothérapie chez les CS ?
essais sytématiques?
avant poussée de croissance pubertaire?
. Nouvel(s) essai(s) chez les CR ?
- au moins un 2ème essai pour conclure à une CR?
- autre(s) essai(s) + tardif(s) ? Avant une GMO?
. Prévention des effets secondaires des CT +++

T. Leblanc 2018
ABD : 4 profils
de réponse à la corticothérapie
Corticosensibles
- « vrais » corticosensibles: doses « homéopathiques »
- corticosensibles médiocres voire à minima…
Corticodépendants à dose élevée: NE PAS INSISTER!
CorticorésistantsAu total :
40% des pts sont dépendants des
transfusions au long cours
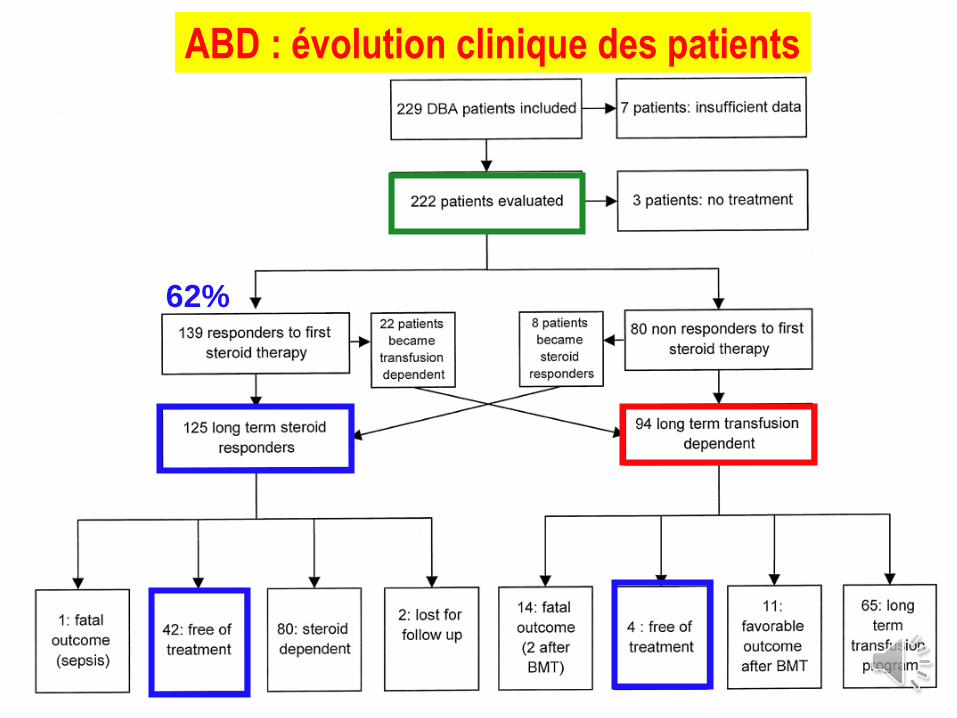
T. Leblanc 2013
ABD : évolution clinique des patients
62%
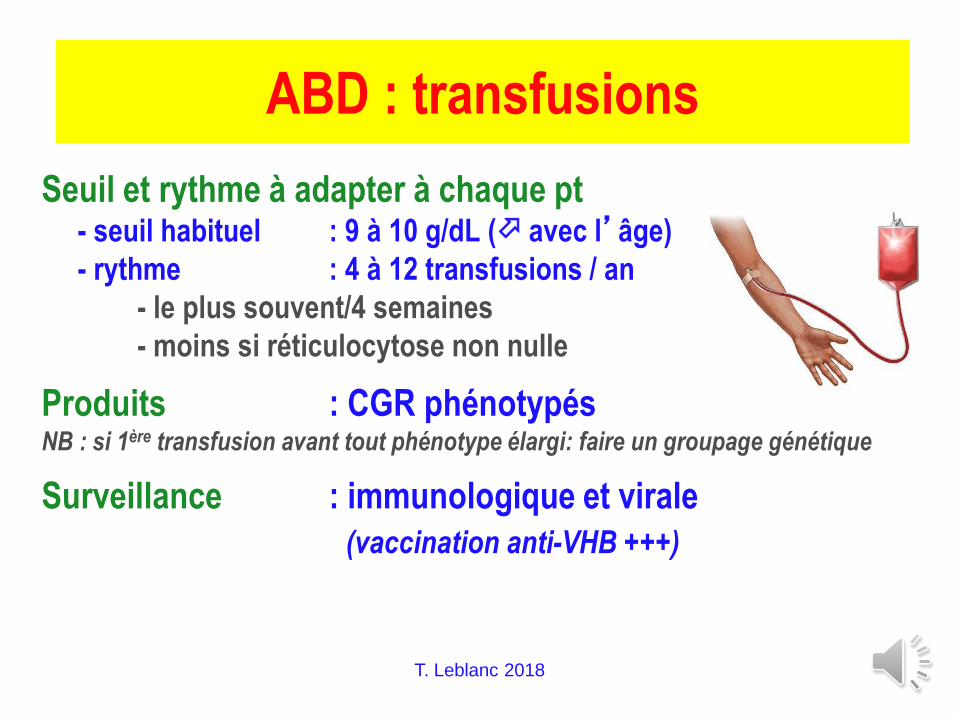
T. Leblanc 2018
ABD : transfusions
Seuil et rythme à adapter à chaque pt- seuil habituel : 9 à 10 g/dL ( avec l’âge)
- rythme : 4 à 12 transfusions / an
- le plus souvent/4 semaines
- moins si réticulocytose non nulle
Produits : CGR phénotypésNB : si 1ère transfusion avant tout phénotype élargi: faire un groupage génétique
Surveillance : immunologique et virale
(vaccination anti-VHB +++)
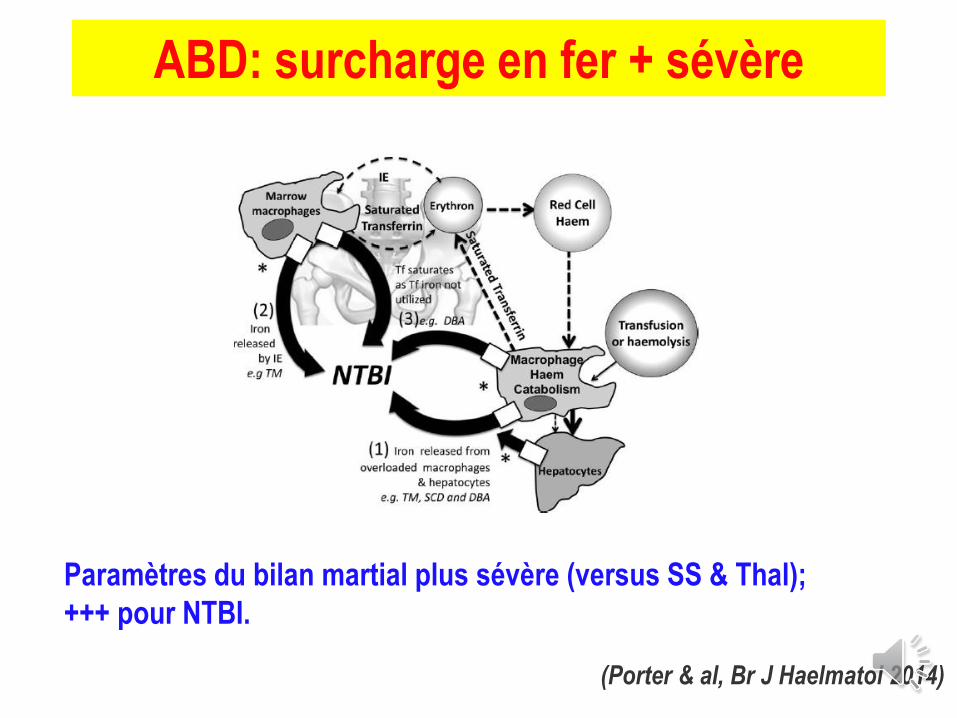
(Porter & al, Br J Haelmatol 2014)
ABD: surcharge en fer + sévère
Paramètres du bilan martial plus sévère (versus SS & Thal);
+++ pour NTBI.

Chélation : modalités (1)
A débuter pour une ferritine «approchant» 1000 mg/l
Deferoxamine ou Deferasirox en 1ère intention
ABD: problème spécifique des tout-petits: - Surcharge en fer très précoce chez les enfants ABD
- Pas d’AMM pour le déférasirox en dessous de 2 ans + acceptabilité
- Déféroxamine + toxique (croissance): ne pas dépasser 30 mg/kg/j
Adaptation des doses: - à discuter au bout de 3 mois
- augmenter par paliers si besoin (+++ Exjade) / discuter association
- ne pas non plus surchélater +++; risque toxique > si ferritine basse
Classiquement:
pas < 500
DFP:
Agranulocytose!

Chélation : modalités (2)
Surveillance +++ :
- efficacité: taux de ferritine, IRM foie & cœur, bilans
endocriniens,…
- toxicité (selon agent) : NFS, BH, BR avec clairance, OPH,
audition,…
Penser à l’adhésion au traitement (AJA +++)
Place des
associations ?

ABD : traitements alternatifs
Bolus de corticoïdes : NON!
Ciclosporine-A : non
IgIV : non
Interleukine 3 : ND
Erythropoiétine : NON
Métoclopramide : ?
Sotatercept : non
Leucine : ?
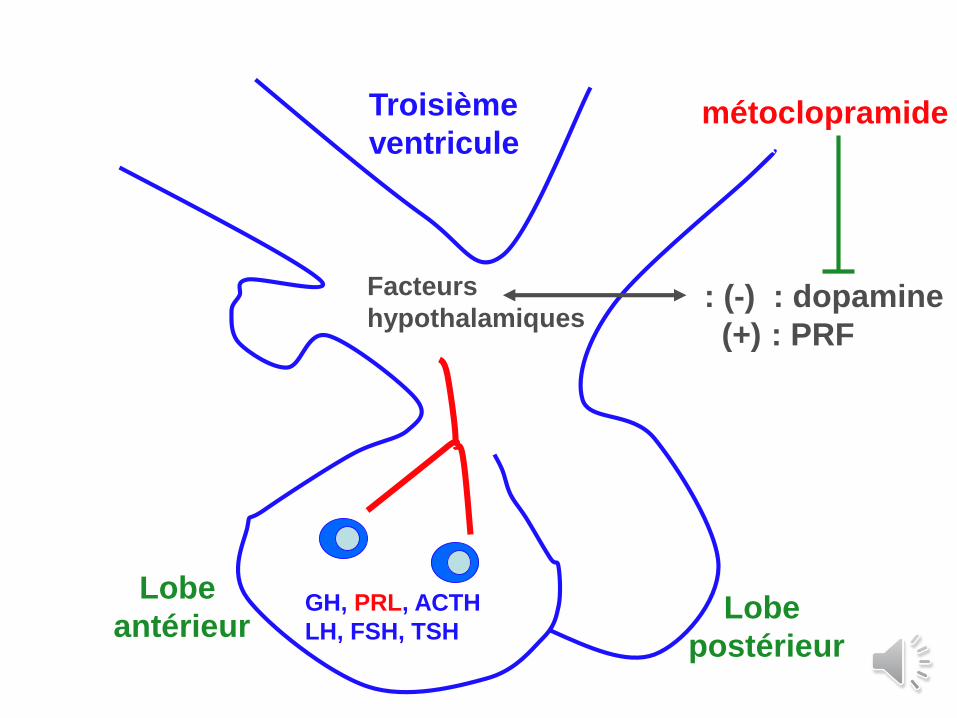
GH, PRL, ACTH
LH, FSH, TSH
Lobe
antérieurLobe
postérieur
Facteurs
hypothalamiques: (-) : dopamine
(+) : PRF
Troisième
ventriculemétoclopramide
0
2
4
6
8
10
12
14
23
/11
/98
23
/02
/99
23
/05
/99
23
/08
/99
23
/11
/99
23
/02
/00
23
/05
/00
23
/08
/00
23
/11
/00
23
/02
/01
23
/05
/01
23
/08
/01
23
/11
/01
23
/02
/02
23
/05
/02
23
/08
/02
23
/11
/02
23
/02
/03
23
/05
/03
23
/08
/03
23
/11
/03
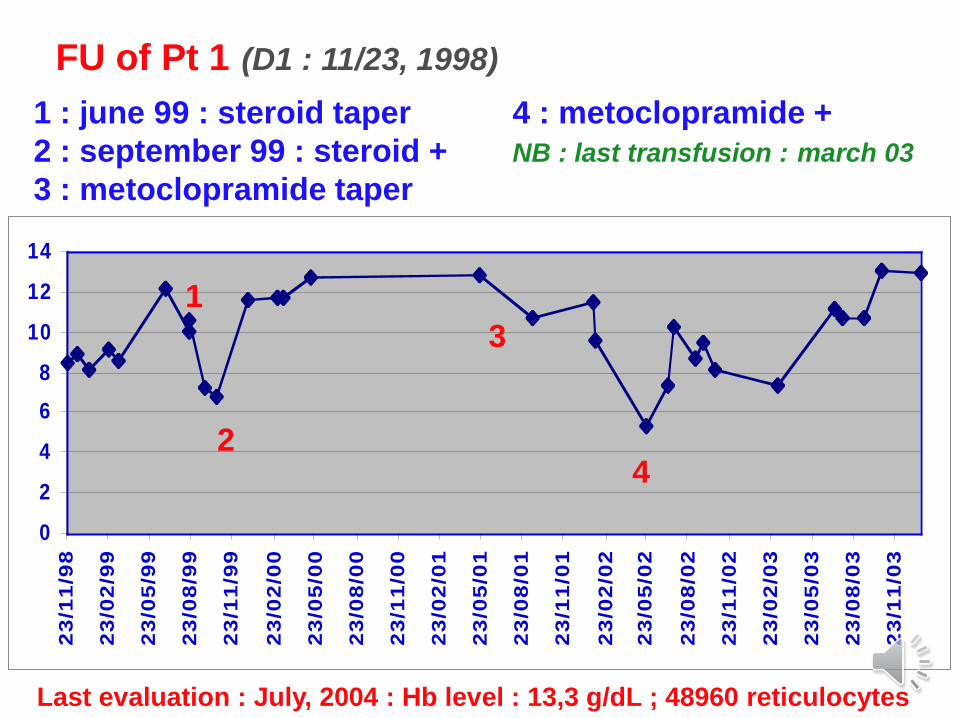
FU of Pt 1 (D1 : 11/23, 1998)
1
2
3
4
1 : june 99 : steroid taper 4 : metoclopramide +
2 : september 99 : steroid + NB : last transfusion : march 03
3 : metoclopramide taper
Last evaluation : July, 2004 : Hb level : 13,3 g/dL ; 48960 reticulocytes

Faut il utiliser la Leucine?
Toujours très peu de données: à ce jour la seule
publication date de 2008 avec un unique enfant en
RP…
Mais:
1) Bénéfice sur EG & croissance
2) essais US en cours.
Toxicité liée à l’usage prolongé de leucine?
Innocuité des préparations disponibles sur internet?
Problème: comment éviter ou, si déjà effective, encadrer l’auto-
prescription…

T. Leblanc 2017
ABD : allogreffe (1)
Déficit historique de greffe dans l’ABD:
- risque inhérent à la greffe
- possibilité de «rémission»
Résultats actuels très améliorés

Allogreffe de moelle: expérience Française
N = 28 (29 greffes)
Période de greffe: < 2000: 6
: 2000 à 2010: 12
: ≥ 2010: 11
Age médian à la greffe: 6,8 ans [1-32]
Type de donneur: moelle +++ : 21 MSD & 5 MUR
Modalités de greffe: classiques: moelle, conditionnement chimioablatif (pas d’ICT)
(Jean-Hugues DALLE)
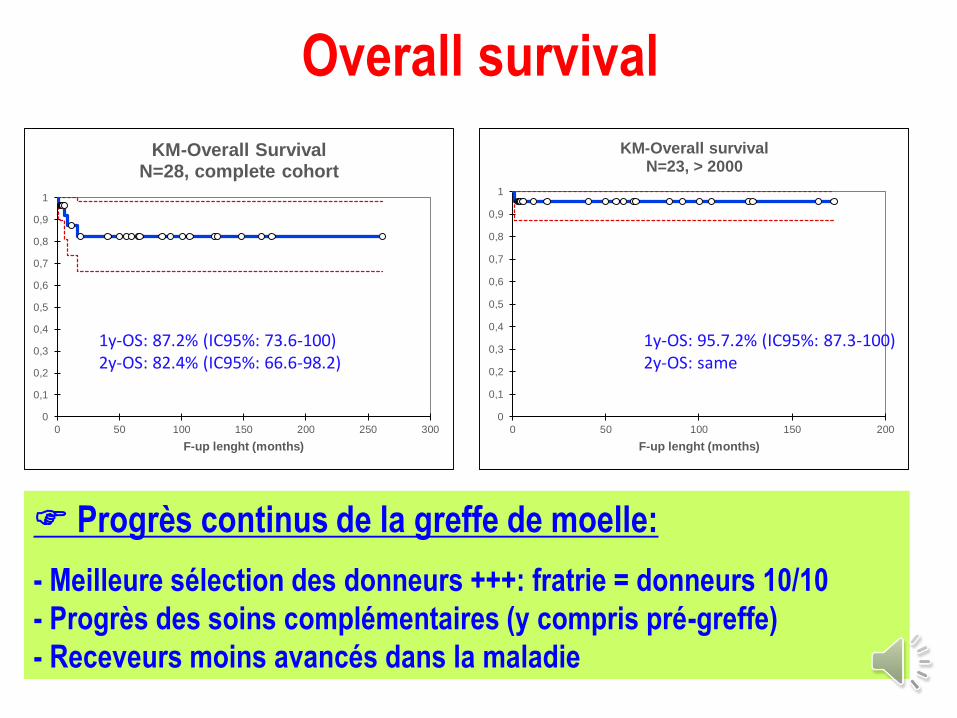
Overall survival
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1
0 50 100 150 200 250 300
F-up lenght (months)
KM-Overall SurvivalN=28, complete cohort
1y-OS: 87.2% (IC95%: 73.6-100)2y-OS: 82.4% (IC95%: 66.6-98.2)
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1
0 50 100 150 200
F-up lenght (months)
KM-Overall survivalN=23, > 2000
1y-OS: 95.7.2% (IC95%: 87.3-100)2y-OS: same
Progrès continus de la greffe de moelle:
- Meilleure sélection des donneurs +++: fratrie = donneurs 10/10
- Progrès des soins complémentaires (y compris pré-greffe)
- Receveurs moins avancés dans la maladie

T. Leblanc 2018
ABD : allogreffe
Consensus actuel pour les indications
1. Uniquement pour les pts transfusés
2. Greffes:
- génoidentique: pb. : exclusion de l’ABD chez un
donneur familial +++
- phénoidentiques si MUD 10/10 (Consensus EuroDBA
Octobre 2017)
3. Greffer tôt (< 5 ans)Mauvais résultats
au-delà de 10 Ans…

ABD : problèmes cliniques à l’âge adulte
• Support transfusionnel au long cours & surcharge
en fer +++
• Toxicité au long cours des corticoïdes
• Développement d’un déficit B de type DCV
• Conseil génétique & grossesse
• Evolutions hématologiques: "rechute", bi ou
tricytopénies & risque d’évolution vers un SMD
• Cancers

T. Leblanc 2018
DBA guidelines
(Br J Haematol. 2008 Sep;142(6):859-76)






























