Embed Size (px)
DESCRIPTION
Introduction à La Pharmacologie
Citation preview

Pr My El Abbes FAOUZI
LABORATROIRE DE PHARMACOLOGIE
ET DE TOXICOLOGIE
FACULTE DE MEDECINE ET DE PHARMACIE
-RABAT-
Introduction à la Pharmacologie
et vie d'un médicament

Le diagnostic des maladies est une étape importante dans
les soins.
La prescription et la délivrance des médicaments sont des
gestes qu’il faut contrôler afin d’en tirer le maximum
d’efficacité du médicament et le minimum de risques.
L’utilisation des médicaments chez les malades a un
double effet, un effet positif : le traitement et un effet
négatif : les effets indésirables.
Les médicaments les plus chers ne sont pas
obligatoirement les plus efficaces.

DEFINITIONS
Un médicament peut être défini comme toute substance ou composition présentés comme possédant des propriétés curatives ou préventives à l’égard des maladies humaines ou animales, ainsi que tout produit pouvant être administré à l’homme ou l’animal, en vue d’établir un diagnostic médical ou restaurer, corriger ou modifier leurs fonctions organiques.
On peut désigner le médicament sous le nom de drogue à ne pas confondre avec les substances toxicomanogènes.
PHARMACOLOGIE
La Pharmacologie est l’étude des médicaments, de leur action et de leur emploi.

Un médicament (ou une médication) est une substance chimiquequi exerce une action bénéfique particulière sur l’organisme(animal ou humain). La science des médicaments est appeléepharmacologie. La prescription de médicaments est lapharmacothérapie. De nombreux médicaments sont d’originevégétale, animale ou biologique, mais la plupart desmédicaments actuels sont des produits de synthèse.
Pharmacocinétique
Discipline qui a pour objet l’étude descriptive et quantitative du devenir du médicament dans l’organisme auquel il est administré.
Mars 2004 : Directive européenne relative à la définition dumédicament.

INTRODUCTION GÉNÉRALE ÉVOLUTION
HISTORIQUE DE LA RECHERCHE PHARMACOLOGIQUE
2000 av. J.-C. Médecine égyptienne antique : utilisation
d’onguents, de produits à inhaler et de pilules avec
opium, graines de lin, canabis.
Antiquité gréco-romaine : plantes et herbes
Moyen-âge : opium comme anesthésique
1500 apr. J.-C. Médecine arabe : remèdes à base de plantes et de
minéraux
Renaissance: connaissance de l’anatomie humaine (Vésale, DaVinci)
XVIIIe - XIXe siècles :
– révolution du microscope
– première vaccination par E. Jenner
– Pasteur : découverte des micro-organismes comme cause d’infection

Importance de l’hygiène et utilisation du phénol comme
antiseptique (Semmel Weiss)
1840 : découverte de l’éther, du chloroforme comme
anesthésiques
• XXe siècle : découverte de nouveaux médicaments : au
début, plutôt des découvertes accidentelles (par ex.
pénicilline par Alexander Fleming)
• Recherche progressivement plus ciblée
Au niveau des organes / tissus
Au niveau des cellules
Au niveau des molécules / récepteurs
L’avenir : biotechnologie & médicaments sur mesure
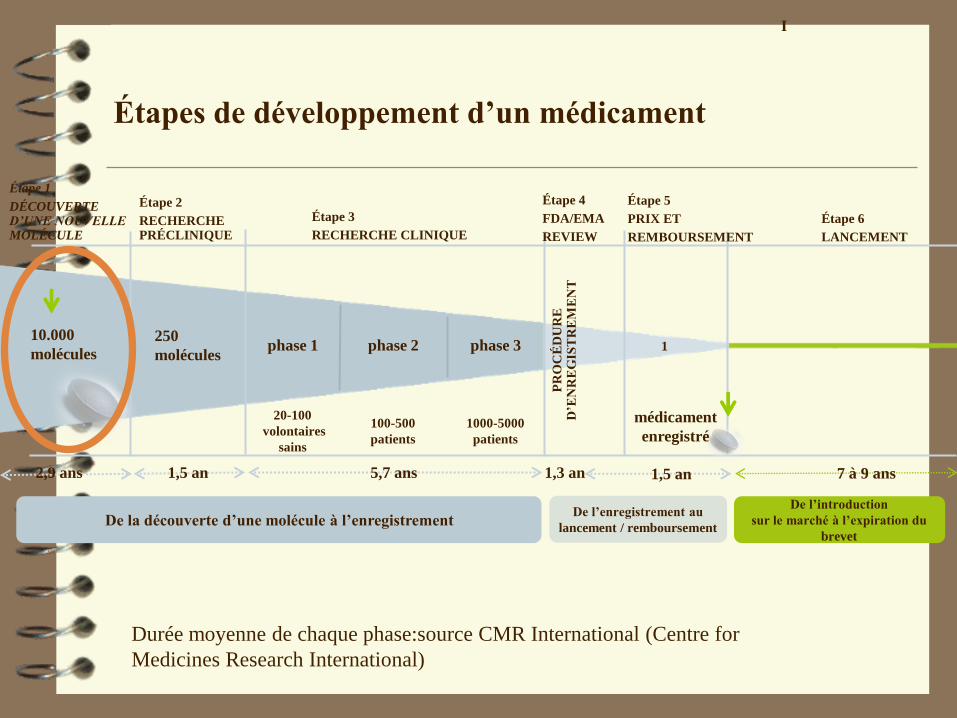
Étapes de développement d’un médicament
Durée moyenne de chaque phase:source CMR International (Centre for
Medicines Research International)
I
10.000
moléculesphase 2phase 1
5,7 ans
1000-5000
patients
100-500
patients
1
médicament
enregistré
phase 3
1,3 an
Étape 5
PRIX ET
REMBOURSEMENT
De l’introduction
sur le marché à l’expiration du
brevet
1,5 an 1,5 an
Étape 6
LANCEMENT
7 à 9 ans
De la découverte d’une molécule à l’enregistrementDe l’enregistrement au
lancement / remboursement
250
molécules
PR
OC
ÉD
UR
ED
’EN
RE
GIS
TR
EM
EN
T
Étape 1
DÉCOUVERTE D’UNE NOUVELLE MOLÉCULE
Étape 2
RECHERCHE PRÉCLINIQUE
Étape 3
RECHERCHE CLINIQUE
Étape 4
FDA/EMA
REVIEW
20-100
volontaires
sains
2,9 ans

III. SCREENING ET SÉLECTION DE NOUVELLES
MOLÉCULES
• Chimistes, pharmacologues et biologistes ont investigué
sur des milliers de molécules existantes ou en ont créé de
nouvelles.
• On produit parfois des milliers de « têtes de série » (de
composés prometteurs). Ces substances sont susceptibles
d’agir sur les mécanismes responsables de certaines
maladies ou affections.
Identification de nouveaux composés
Balayage de « bibliothèques » de composés
Production de nouveaux composés

V. NOUVEAUX DÉVELOPPEMENTS DANS LA RECHERCHE
LA BIOTECHNOLOGIE MODERNE : MOMENTS HISTORIQUES
1675 : Découverte des micro-organismes
1856 : G. Mendel découvre les lois de l’hérédité
1953 : James Watson et Francis Crick décrivent la structure de l’ADN
1975 : Développement d’une technique permettant de produire des
anticorps grâce à la biotechnologie
1980 : La biotechnologie moderne utilise la technologie de
recombinaison de l’ADN. Le micro-organisme E. Coli est utilisé pour
produire de l’insuline humaine.
1982 : Premier médicament recombinant disponible sur le marché
2001 : Le « Human Genome Project » est terminé

Brevet = l’acquisition d’un droit exclusif sur la production,
l’utilisation, la vente, l’accord de licence, et l’importation de
l’invention brevetée
Quasi immédiatement après la découverte d’une molécule
potentiellement adéquate
DEMANDE DE BREVET
Le brevet, un incitant à la créativité
• Conditions:
Nouveau = innovateur
Inventif
Applicable à l’industrie
Brevet = garantir l’innovation
I

Quoi ?
Les molécules sélectionnées (résultat du screening)
Indications
Formules
Combien de temps ?
Le brevet reste valide pendant 20 ans (à ce stade, la
recherche, qui prend environ 10-15 ans, doit encore
commencer)
Exception: prolongation pour 5 ans
Par qui ?
Au niveau international
European Patent Office,
Au niveau national
DEMANDE DE BREVET
I

RECHERCHE PRECLINIQUERésumé
I. Qu’est-ce que la recherche préclinique ?
II. Toxicologie: toxicité aiguë
III. Toxicologie: toxicité chronique
IV. Pharmacologie
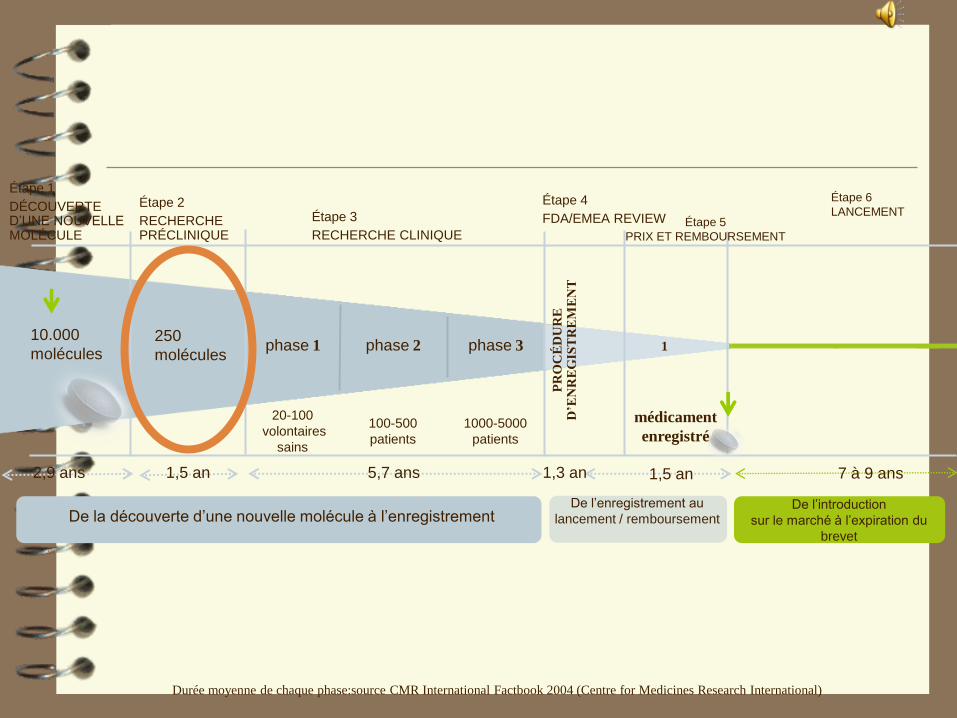
Durée moyenne de chaque phase:source CMR International Factbook 2004 (Centre for Medicines Research International)
10.000
moléculesphase 2phase 1
5,7 ans
1000-5000
patients
100-500
patients
1
médicament
enregistré
phase 3
1,3 an
Étape 5
PRIX ET REMBOURSEMENT
De l’introduction
sur le marché à l’expiration du
brevet
1,5 an 1,5 an
Étape 6
LANCEMENT
7 à 9 ans
De la découverte d’une nouvelle molécule à l’enregistrementDe l’enregistrement au
lancement / remboursement
250
molécules
PR
OC
ÉD
UR
ED
’EN
RE
GIS
TR
EM
EN
T
Étape 1
DÉCOUVERTE D’UNE NOUVELLE MOLÉCULE
Étape 2
RECHERCHE PRÉCLINIQUE
Étape 3
RECHERCHE CLINIQUE
Étape 4
FDA/EMEA REVIEW
20-100
volontaires
sains
2,9 ans

I- QU’EST-CE QUE LA RECHERCHE PRECLINIQUE ?
Tests de médicaments brevetés sur
– cultures de cellules (in vitro)
– animaux (in vivo)
Recherche de
– Sécurité et toxicité: en vue de l’utiliser plus tard chez les humains
– Aspect chimique: pureté, stabilité, ...
– Mécanismes d’action possibles: le médicament rencontre tout à fait les besoins définis pendant la recherche fondamentale (au niveau moléculaire)
– Aspects pharmaceutiques: formulation (comprimés, gélules…)
– Aspects pharmacocinétiques: absorption, métabolisation ...
– Production: possibilité technique de production à grande échelle

II. TOXICOLOGIE: TOXICITÉ AIGUË
Administration d’une dose unique ou de plusieurs
doses d’un médicament
Enregistrement des effets toxiques à court terme
prédiction de la toxicité et des effets indésirables chez l’homme
Définition de
– la dose létale (DL50)
– la dose maximale tolérée (MTD)
Lésions aiguës des organes, tissus, cellules…

III. TOXICOLOGIE: TOXICITÉ CHRONIQUE
Administration à long terme (mois-années) et répétée d’un
médicament et enregistrement d’effets toxiques
– Effet d’une exposition de longue durée sur les organes,
les tissus, les cellules
Etudes ciblées:
– Tératogenèse Mutagenèse
– Génotoxicité Oncogenèse
– Tolérance locale Reproduction

IV. PHARMACOLOGIE
Pharmacocinétique
– Comment un produit est-il assimilé et
transformé par l’organisme?
Pharmacodynamie
– Quel effet a un produit sur l’organisme?

LA RECHERCHE CLINIQUE
RESUME
I. Qu’est-ce qu’un programme de recherche clinique ?
II. Les différentes phases en recherche clinique
III. Qu’est-ce qu’une étude clinique ?
I. L’étude de référence: l’étude contrôlée randomisée
II. Signification de placebo, randomisation, aveugle
IV. Le traitement des résultats de l’étude
V. L’interprétation des résultats d’une étude clinique
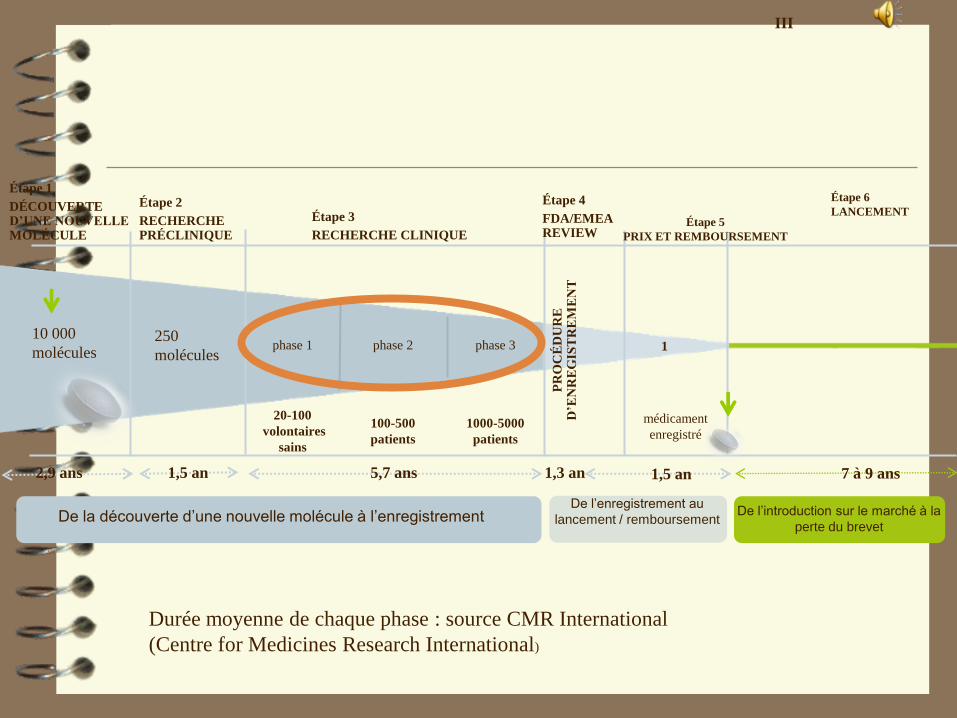
Durée moyenne de chaque phase : source CMR International
(Centre for Medicines Research International)
10 000
moléculesphase 2phase 1
5,7 ans
1000-5000
patients
100-500
patients
1
médicament
enregistré
phase 3
1,3 an
Étape 5
PRIX ET REMBOURSEMENT
De l’introduction sur le marché à la
perte du brevet
1,5 an 1,5 an
Étape 6
LANCEMENT
7 à 9 ans
De la découverte d’une nouvelle molécule à l’enregistrementDe l’enregistrement au
lancement / remboursement
250
molécules
PR
OC
ÉD
UR
ED
’EN
RE
GIS
TR
EM
EN
T
Étape 1
DÉCOUVERTE D’UNE NOUVELLE MOLÉCULE
Étape 2
RECHERCHE PRÉCLINIQUE
Étape 3
RECHERCHE CLINIQUE
Étape 4
FDA/EMEA REVIEW
20-100
volontaires
sains
III
2,9 ans

I. QU’EST-CE QU’UN PROGRAMME DE RECHERCHE CLINIQUE ?
Ensemble d’études cliniques où l’efficacité et la sécurité
d’un médicament sont étudiées chez l’homme
Les études se déroulent sur plusieurs phases :
– Études de phase I
– Études de phase II
– Études de phase III
– (Études de phase IV: après la mise sur le marché d’un
médicament)

QU’EST-CE QU’UN PROGRAMME DE RECHERCHE CLINIQUE ?
Ensemble d’études conçues en collaboration avec des spécialistes
médicaux et les autorités (EMA/FDA) dans lesquelles les éléments
suivants sont définis :
Objectifs des études: ce que l’on va étudier
Durée : combien de temps les études doivent-elles durer pour être pertinentes ?
Le matériel de comparaison : comparaison avec un placebo ou avec un autre médicament
Ce que l’on considère comme étant un résultat concluant, quelles sont les méthodes « de mesure » que l’on va utiliser
Aspects de sécurité : par ex. attention particulière à la sécurité sur le plan cardiologique
Quels groupes de patients doit-on examiner (fumeurs, + de 65ans, femmes…) ?
Quelles sont les études d’interaction à mener ?

Participants par étude : entre 20 et 100 volontaires sains (H/F)
Objectif : la sécurité avant tout
– Définition d’un profil de sécurité
– Définition d’un dosage sans risque et de marges de sécurité
– Pharmacocinétique (et pharmacodynamique)
– En général, pas de recherche sur l’efficacité
Localisation
Principalement en milieu hospitalier (dans un contexte rigoureux)
Durée : environ 18 mois
Budget : 5 % du coût total de développement = 46 millions de dollars US ou 37
millions d’euros
Une nouvelle substance a ± 60 % de chances de survivre à cette phase
PHASE I : ÉTUDES MENÉES SUR DES VOLONTAIRES SAINS
II. LES DIFFÉRENTES PHASES EN RECHERCHE CLINIQUE

Participants : 100 à 500 patients (H/F)
– Profil défini dans le programme de développement
– Les études sont proposées au patient par le médecin traitant
Localisation : généralement en milieu hospitalier
– Cadre strictement règlementé, sous contrôle médical
Objectif
– Etude de l’efficacité et approfondissement de la recherche en matière de
sécurité (toxicologie, effets secondaires)
– Définition d’un premier schéma de dosage
– Approfondissement de la recherche sur les plans pharmacocinétique et
pharmacodynamique
PHASE II : ÉTUDES MENÉES SUR DE PETITS GROUPES DE PATIENTS
II. LES DIFFÉRENTES PHASES EN RECHERCHE CLINIQUE

PHASE II : ÉTUDES MENÉES SUR DE PETITS GROUPES DE PATIENTS
La substance exerce-t-elle l’action escomptée (par ex. faire diminuer la tension
artérielle) ?
Le modèle théorique correspond-il à la réalité ?
II. LES DIFFÉRENTES PHASES EN RECHERCHE CLINIQUE
Durée : environ 18 mois
Budget : 10 % du coût de développement total, soit 85
millions de dollars US ou 70 millions d’euros
Évaluation « preuve du concept »
III

PHASE III : ÉTUDES MENÉES SUR DES GROUPES DE PATIENTS PLUS
IMPORTANTS (POPULATION « REAL LIFE »)
Participants
de 500 à 10.000 patients
Localisation
En milieu hospitalier et en ambulatoire
Durée
– De 2,5 à 4 ans (jusqu’à 7 années
parfois)
Budget
– 11 % du coût total de développement,
soit 102 millions de dollars US ou 84
millions d’euros
• Objectif
Nouvelle confirmation des
points suivants
Efficacité
Sécurité
Etude des effets secondaires
Collecte de données
statistiques visant à
confirmer les résultats de la
phase II
Données destinées aux
dossiers d’enregistrement
II. LES DIFFÉRENTES PHASES EN RECHERCHE CLINIQUE

III. QU’EST-CE QU’UNE ÉTUDE CLINIQUE ?
Quelques définitions :
Définition de la recherche clinique: acquisition de connaissances
sur une maladie et/ou un traitement, une nouvelle technique de
diagnostic, instruments médicaux…chez l’homme, d’une manière
éthiquement acceptable. Ceci peut également être de la recherche
épidémiologique.
Définition d’une étude clinique : forme de recherche scientifique
dans laquelle l’efficacité, la sécurité et d’autres aspects d’un ou de
plusieurs traitements sont étudiés. Constitue l’un des éléments d’un
programme de recherche clinique.
III

III
III. QU’EST-CE QU’UNE ÉTUDE CLINIQUE ?
Bon à savoir :
Toute étude se déroule sous surveillance médicale stricte (dans un
environnement spécialisé, hôpital, cabinet du médecin traitant...)
Avant de pouvoir commencer, une étude doit d’abord être
approuvée par un comité d’éthique.
Pour chaque étude, des directives internationales sont suivies pour
garantir la sécurité du participant
Chaque personne participant à une étude le fait de manière
volontaire et peut à tout moment arrêter sa participation sans
donner de raison
Il existe différents types d’études avec différents designs possibles
En moyenne 30 à 80 études réalisées avant l’enregistrement
III

III. QU’EST-CE QU’UNE ÉTUDE CLINIQUE EXACTEMENT ?
Eléments fondamentaux :
Chaque étude se passe selon un schéma défini à l’avance, selon un
protocole
– Chaque étude a son propre protocole
Toutes les données sont minutieusement notées dans un CRF (Case
Report Form), à savoir un cahier préimprimé dans lequel toutes les
données du patient sont tenues à jour de façon anonyme.
– Dans une étude, chaque patient a son propre CRF
– Toutes les données médicales collectées par prélèvements sanguins,
radios, examens cliniques… y sont consignées
– Toutes les prises de médicaments sont notées exactement
– Tous les effets secondaires sont consignés
– Tous les résultats sont objectivement notés (p.ex. : amélioration de
la douleur sur une échelle de douleur)

III. QU’EST-CE QU’UNE ÉTUDE CLINIQUE EXACTEMENT ?
L’ÉTUDE RANDOMISÉE CONTRÔLÉE
(RCT = RANDOMISED CONTROLED TRIAL)
Les participants sont sélectionnés sur la base du protocole : critères
d’inclusion et d’exclusion
Les participants sont informés et donnent leur accord : Informed
Consent
Approbation de l’étude par un Comité Éthique
Randomisation et notion d’aveugle
Prospective
Deux groupes : exemple le plus simple : le groupe A reçoit le principe
actif et le groupe B reçoit un placebo ou une médication de référence
III

III
« le placebo » : médication sans principe actif
Effet placebo: effet (résultat) obtenu après administration d’un
placebo ou un autre traitement qui, en principe, ne devrait pas
avoir d’effet
Le groupe placebo: le groupe de patients qui reçoit dans une
étude « le placebo »
III. QU’EST-CE QU’UNE ÉTUDE CLINIQUE EXACTEMENT ?
Signification de « PLACEBO »
III

Contrôle par placebo dans une étude : administration d’une
substance inactive à un groupe de patients de contrôle. On
compare les résultats enregistrés dans le groupe « placebo » avec
ceux enregistrés dans le groupe « actif ».
III. QU’EST-CE QU’UNE ÉTUDE CLINIQUE
EXACTEMENT ?
Signification de « PLACEBO »
III
Groupe de patients
recevant le placebo
Groupe de patients
recevant le principe actif

III. QU’EST-CE QU’UNE ÉTUDE CLINIQUE EXACTEMENT ?
La notion « d’AVEUGLE »
Signification de « RANDOMISATION »
III
Simple
aveugleouvert
Double
aveugle
Groupe traité Groupe de contrôle

L’ensemble des données de l’étude est introduit dans des bases
de données :
– De manière complètement anonyme
– Suivi d’une analyse statistique
Présentation des premiers résultats dans le cadre de congrès
internationaux
Publications
Méta-analyses
IV. LE TRAITEMENT DES RÉSULTATS DE L’ÉTUDE
III

III
Les résultats d’une étude ne peuvent jamais être considérés
isolément. Ils s’inscrivent dans un cadre plus large.
Un résultat ne peut jamais être interprété tel quel
– doit toujours être comparé
– doit toujours être placé dans un contexte plus large
Comparaison avec d’autres études
V. INTERPRÉTATION DES RÉSULTATS D’ÉTUDES CLINIQUES
III

Le titre > objet, but de l’étude
Les auteurs > valeur, sérieux de l’étude
L’introduction > permet de placer l’étude dans un contexte plus
large
Méthodologie > comment et de quelle manière les résultats ont été
obtenus et mesurés, comparabilité des données
Schéma de l’étude & critères d’évaluation > prédéfinis
Méthode statistique utilisée > également prédéfinie
Les résultats > pas de différence statistique ou différence
statistique (significative ou non : valeur P)
La discussion > résultats mis en perspective
Les conclusions > résultats examinés et interprétés dans le contexte
global
Références avec date de publication
V. INTERPRÉTATION DES RÉSULTATS D’ÉTUDES CLINIQUES :
ÉLÉMENTS CLÉS

Question clé : comment déterminer qu’un écart entre deux
groupes est imputable à un traitement spécifique et non au
hasard ?
Risque : risque d’attribuer une différence à la thérapie alors
qu’elle est le fait du hasard.
La valeur P indique le caractère « significatif » d’un écart, à
savoir le fait qu’un écart relevé entre deux groupes n’est pas
uniquement imputable au hasard (P < 0,05 = écart significatif).
– P = 0,05 : 1 chance sur 20 que l’écart soit encore dû au
hasard.
– P = 0,01 : 1 chance sur 100 que l’écart soit dû au hasard.
V. INTERPRÉTATION DES RÉSULTATS D’ÉTUDES CLINIQUES :
NOTIONS STATISTIQUES
III

Qu’est-ce qu’une étude d’économie de la santé ?
– Une comparaison entre différentes options thérapeutiques
(p. ex. traiter avec le produit A, avec le produit B, ne rien
faire ou opérer), dans laquelle on observe le résultat final
non seulement en termes de santé, mais aussi en termes de
coûts
– Question : quelle option fournit le plus grand gain de santé
par unité de capital investi?
– En tenant compte des coûts réels qu’entraîne une maladie :
hospitalisation / consultations médecin traitant / congé de
maladie …
VI. ÉTUDES D’ÉCONOMIE DE LA SANTÉ ou: Quelle valeur la
santé a-t-elle ? Les Finances ont des limites, les besoins pas.
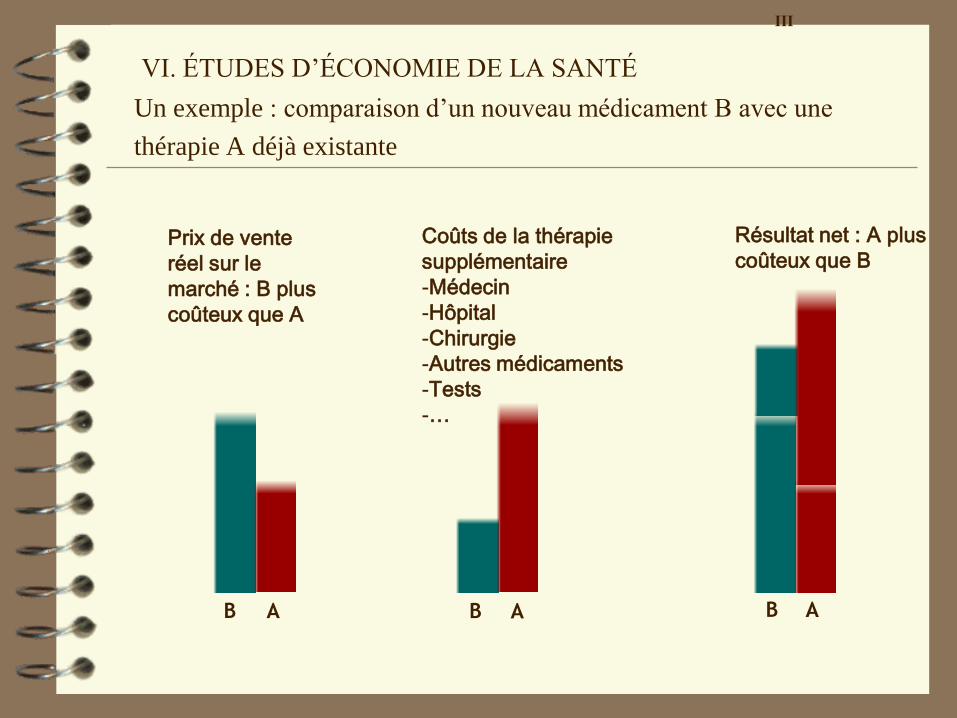
VI. ÉTUDES D’ÉCONOMIE DE LA SANTÉ
Un exemple : comparaison d’un nouveau médicament B avec une
thérapie A déjà existante
B A
Prix de vente
réel sur le
marché : B plus
coûteux que A
Coûts de la thérapie
supplémentaire
-Médecin
-Hôpital
-Chirurgie
-Autres médicaments
-Tests
-…
B A
Résultat net : A plus
coûteux que B
B A
III

CLASSIFICATION DES MEDICAMENTS
1-Les médicaments préventifs
Ils sont administrés au sujet sain, en vue de le protéger contre
une maladie future (vaccins) ou de modifier temporairement un
processus physiologique.
2-Les médicaments substitutifs
Ils pallient une carence de l’organisme, qui peut être d’origine
exogène (alimentaire, vitamines) ou endogène (hormone, facteurs de
coagulations), définitive (insuffisance de la production d’insuline
chez le diabétique) ou provisoire (insuffisance de la production de
progestérone au cours de la gestation)

3-Les médicaments curatifs :
Ils s’attaquent à la cause même de l’état pathologique et permettent d’obtenir la guérison du malade. Ce traitement n’est actuellement possible que pour les maladies infectieuses et parasitaires. Les substance actives entraînent soit la mort de l’agent responsable (produits bactéricides), soit le ralentissement de sa multiplication (produits bactériostatiques) pour permettre aux défenses naturelles de détruire les germes survivants.
4-Les médicaments symptomatiques
Ils sont administrés pour atténuer les symptômes qui résultent d’un état pathologiques (douleur, inflammation, épilepsie, fièvres…) sans qu’ils s’attaquent à la cause même de celui-ci.
Ils atténuent, stimulent ou inhibent le fonctionnent d’un organe. L’administration de ces médicaments doit être prolongée aussi longtemps que la cause n’est pas éliminée.
Ces médicaments sont nombreux (analgésiques, antipyrétiques ; hypnotiques, anti-hypertenseurs etc…)

DÉNOMINATION DES MÉDICAMENTS
I- Dénomination scientifique :
La dénomination des substances chimiques suit les règles de la nomenclature internationale.
II- Dénomination commerciale :
Il s’agit de nom de marque ou de nom commercial faisant l’objet de marques déposées, propriétés de personnes ou de firmes pharmaceutiques.
III- Dénomination commune internationale :
Afin de pallier aux inconvénients respectifs présentés par les dénominations scientifiques et commerciales, l’OMS adopta des directives générales pour l’attribution des Dénominations Communes Internationales (DCI).
La DCI permet d’attribuer à chaque principe actif utilisé en thérapeutique un nom simple et utilisable dans tous les pays, elle permet également de reconnaître la famille à laquelle appartient le médicament.





























