Embed Size (px)
Citation preview

NITROXYDES-LXX1
SYNTHESE DE DERIVES NITROXYDES DU TROPANE.
ETUDE PAR RPE ET RMN DES INTERACTIONS HYPERFINES A LONGUE DISTANCE DANS CES RADICAUX
A. RASSAT* et J. RONZAUD Laboratoire de Chimie Organique Physique, Equipe de Recherche Associee au CNRS No. 20,
Departement de Recherche Fondamentale, Centre d’Etudes Nucleaires de Grenoble, B.P. 85, F 38041 Grenoble Cedex, France
(Receiced in France 2 May 1975; Receivedin fhe UKforpublication 26 Augusf 1975)
Rbumk-On prepare la nortropinone a partir de la tropinone (par N-demethylation au chloroformiate d’ethyle et hydrolyse basique du carbamate) et la dimethyl-1,5 nortropinone par une reaction de Schopf-Robinson. Les radicaux libres nitroxydes, derives de ces amines par oxydation, sont stables. Ils presentent des couplages fofts electron-proton a longue distance que I’on mesure par RPE et RMN, et que I’on attribue par deuttriation selective.
Abstract--Tropinone has been converted into nortropinone. using ethyl chloroformate and base hydrolysis. 1,5-Dimethyl nortropinone has been prepared by a Schopf-Robinson reaction. The nitroxides, obtained by oxidation of these amines, exhibit long-range proton hyperhne coupling, which are measured by ESR and NMR and assigned through selective proton-deuterium exchange.
Dans les radicaux libres, les couplages a longue distance sont generalement inferieurs a IG, sauf darts le cas des radicaux a structure bicyclique oti ils peuvent parfois atteindre des valeurs beaucoup plus Clevees.’ Nous avons publk? une etude theorique montrant la stereoselectivite, en grandeur et en signe, des couplages y a longue distance dans deux radicaux bicycliques nitroxydes derives du tropane. Nous presentons ici la preparation de ces composes ainsi que l’etude detaillee, par resonance paramagnetique Clectronique (RPE) et par resonance magnetique nucleaire (RMN), qui a fourni les bases experimentales a notre etude theorique.
Synthke de la nortropinone, de la dimithyl-I,5 nor- tropinone et des radicaux nitroxydes d&icis
La tropinone 1 est demethylee par le chloroformiate d’ethyle.” On obtient facilement le N-Cthylformiate de nortropinone 2 qui ne s’hydrolyse pas en milieu acide’ mais que nous avons reussi a hydrolyser en milieu trts fortement basique, a chaud (75°C). La decarboxylation conduisant a la nortropinone 3a se fait ensuite en milieu acide. La structure de 3a ainsi obtenue a ete confirmee par sa transformation en N-tthylformiate 2 puis en tropine 4 et pseudotropine S..’
Nous inspirant d’un brevet” qui decrit la synthese de la dimethyl-I,5 tropinone 8a, nous avons prepare la dimethyl-1,5 nortropinone 8b en condensant I’hexane dione-2,5 6, I’ammoniaque (sous forme de chlorure d’ammonium) et l’acide acetone-dicarboxylique 7 par la reaction de Schbpf-Robinson.>
Nous n’avons pas rtussi a preparer I’amine secondaire 8b par N-demethylation de 8a au chloroformiate d’ethyle.
L’oxydation des amines secondaires 3a et 8b par I’eau oxygenee en presence d’acide phosphotungstique’ con- duit aux radicaux nortropinone N-oxyle 3b et dimethyl- I,5 nortropinone N-oxyle tk.
RPE des radicaux 3b et 8c ci I’Hat solide
Le radical nitroxyde 3b est un solide jaune (F = 89T), peu soluble. L’intensite de son signal de RPE, quand il est
sous forme de poudre, est beaucoup plus faible (50% environ) que celle d’un tchantillon d’un radical temoin, le tetramethyl-2,2,6,6 pipkridinone4 N-oxyle ou Tano, contenant le m&me nombre de moltcules. En solution a 20°C. nous avons constatt qu’a partir d’une concentration de I’ordre de IM, I’intensite de son signal de RPE est legerement plus faible que celle du signal du Tano pris A la meme concentration. Cet &art entre les intensites augmente avec la concentration. Un radical de structure voisine, le norpseudopelletierine N-oxyle9 est, comme le radical 3b, jaune a l’etat solide, alors que la plupart des radicaux nitroxydes sont rouge-orange. Ce radical est dim&i& a I’etat solide,‘O” les groupements nitroxydes &ant sit&s selon le Schema 3. L’ttat fondamental de ce dimtre est un Ctat singulet, l’tcart singulet-triplet ttant de 5000°K.“” Par analogie, nous pensons que le radical 3b est sous forme de dimere a l’etat solide. Le faible signal de RPE de ce radical en poudre correspondrait done aux dimtres dans I’etat triplet peuple thermiquement”b et a quelques monoradicaux isoles darts des defauts du solide. La variation de I’intensite du spectre de RPE de ce radical
Y3 CooEt R I I
Schema 1. Synthese de la nortropinone (R = H).
239

240 A. RASSAT et I. ROXZXD
0 6 0 1
Schema 2. Synthese de la dimithyl-I,5 nortropinone (R = H).
I am,
R- b:H c:o*
?/
\
Schema 3.
en solution, en fonction de la concentration, est compati- ble avec un Cquilibre monomere-dim&e.‘*
Le radical nitroxyde tk est un solide orange, trts soluble. Son point de fusion (F = 44”5 C) est beaucoup plus faible que celui du radical 3b. A l’etat solide comme en solution, I’intensite de son signal de RPE a 20°C est Bgale a celle du Tano, compost paramagnetique parfait, etudie dans les memes conditions, A temperature ordinaire, ce radical a done un comportement de solide paramagnetique normal. Cependant, il existe une grande analogie entre la structure aux rayons X du radical tk”* et celle du no~seudopelletierine N-oxyle;‘“” les molecules sont encore appariees dans Ie cristal, selon fe Schema 3, mais la distance entre les deux groupements nitroxydes est passee de 2.4 a 3.4 A. Des mesures magnetiques montrent que ce dimere a aussi un itat fondamental singulet, mais I’icart singulet-triplet n’est plus que de 260°K.” 11 est done vraisemblable que les groupements methyles en t&e de pont creent un encombrement sttrique tel que les groupements nitroxydes n’interagis- sent plus que trts faiblement.
RPE des radicaux 3b ef fk en solution Les effets de solva# et les effets de sel” sur la valeur
du couplage electron-azote dans les radicaux nitroxydes sont bien connus. Dans Ie cas des radicaux 3b et tk, nous obtenons Ies meilleurs spectres en utilisant l’eau saturee en chlorure de lithium et une concentration en radical de M/500. Nous interpretons ces spectres (Fig. I) B I’aide des couplages hyperfins suivants: radical 3b: as = 20.25 G; aH = J-75 G (2H); aH = 2.5G (2H); aH = 1~25G (2H); aH = 0.2 G (2H) et a” = 0.1 G (2H); radical &: aN = 19.5 G; aH = 2.5 G (2H); a,, = 1.2 G (2H). Une reproduction des spectres sur ordinateur confirme ces attributions.
En comparant la structure des radicaux 3b et & et l’ensemble des resultats obtenus par I’analyse des spectres de IWE, nous attribuons, sans ambiguite,‘* le coupiage de 575G aux protons en tete de pont du no~ropinone nitroxyde (protons /3). Tous les autres couplages observes sont done des couplages entre I’electron non apparie et des protons ‘y, done des
I
J=t.
Fig. 1. Spectres de RPE des radicaux 3b (a) et Rc fb) en solution M/500 dans l’eau saturke au LiCI. L’Cchelle (+I est de 5 G. Sur la Figure a, nous prkentons aussi un agrandissem~nt fichelle IOOG) du sommet de la structure hype&e Ia plus intense; cet a~andissement montre ies structures dues aux couplages de 0.2 G
etde@lG.
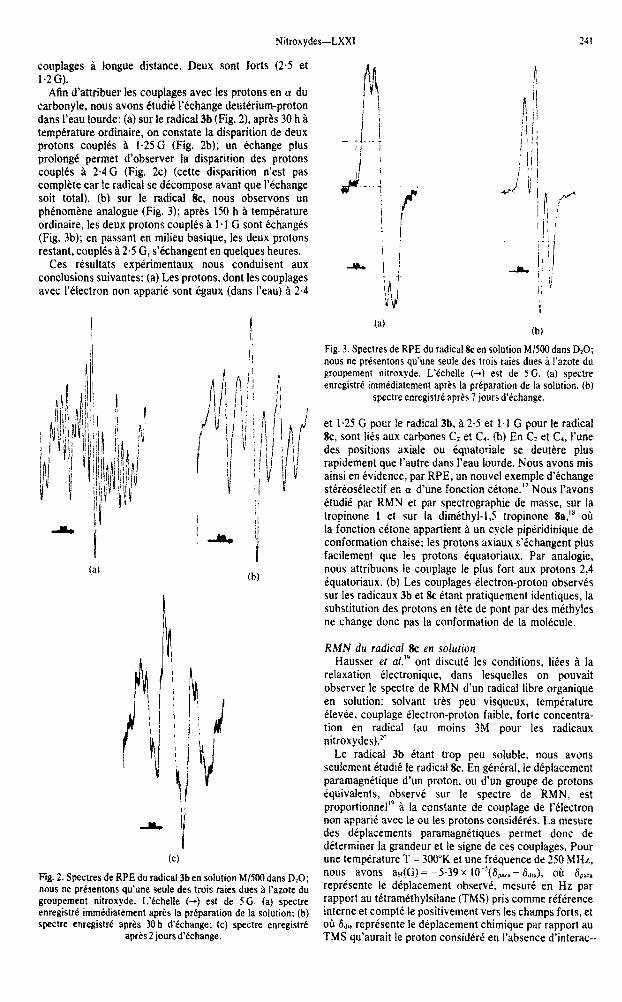
Nitroxydes-LXX1 241
couplages a longue distance. Deux sont forts (2.5 et 1.2G).
Afin d’attribuer les couplages avec les protons en a du carbonyle, nous avons etudie i’echange deut~rium-proton darts I’eau lourde: (a) SW Ie radical 3b (Fig. 2), apres 30 h a temperature ordinaire, on constate la disparition de deux protons couplts a i-25 G (Fig. 2b); un echange plus prolong6 permet d’observer la disparition des protons couples a 2.4 G (Fig. 2c) (cette disparition n’est pas complete car le radical se decompose avant que Y&change soit total). (b) sur le radical &, nous observons un phenomene analogue (Fig. 3); apres 150 h a temperature ordinaire, les deux protons couples a 1.1 G sont &changes (Fig. 3b); en passant en milieu basique, les deux protons restant, couples B 2.5 G, s’echangent en quelques heures.
Ces resultats expkimentaux nous conduisent aux conclusions suivantes: (a) Les protons, dont les couplages avec I’electron non apparii sont egaux (dans I’eau) a 2.4
II\ RMN du radical 8c en solution Hausser et al.lY ont discute les conditions, likes ii la
relaxation Clectronique, dans lesquelles on pouvait observer le spectre de RMN d’un radical libre organique en solution: solvant trts peu visqueux, temp&rature elevee, couplage electron-proton faible, forte concentra- tion en radical (au moins 3M pour les radicaux nitroxydes).*”
w Fig. 2. Spectres de RPE du radical 3b en solution Ml500 dans D?O; now ne prtsentons qu’une seule des trois raies dues B l’azote du
groupement nitroxyde. Lkhelle (-+) est de SG. (a) spectre enregistre immediatement apres la preparation de la solution: (b)
spectre enregistre aprts 30 h d’echange; tc) spectre enregistrt
apres 2 jours d’tchange.
i Ii/
Fig. 3. Spectres de RPE du radical tk en solution M/500 dam D,O; nous ne presentons qu’une seule des trois raies dues ri I’azote du
groupement nitroxyde. L’echelle (-+) est de 5G. (a) spectre
enregistre immtdiatement aprts la preparation de la solution. (b)
spectre enregistre apres 7 jours d’tchange.
et i .25 G pour le radical 3b, h 2-5 et 1. I G pour le radical &, sont lies aux carbones CI et Cd. (b) En Cz et CA, l’une des positions axiale ou Cquatoriale se deutere plus rapidement que l’autre dans l’eau lourde. Nous avons mis ainsi en evidence, par RPE, un nouvel exemple d&change sttreoselectif en a dune fonction c&one.” Nous l’avons Ctudie par RMN et par spectrographic de masse, sur la tropinone 1 et sur la dimithyl-I,5 tropinone 8a,‘” ou la fonction &tone appartient a un cycle piperidinique de conformation chaise; ies protons axiaux s’echangent plus facilement que les protons Cquatoriaux. Par analogie, nous attribuons le couplage le plus fort aux protons 2.4 equatoriaux. (b) Les couplages electron-proton observes sur les radicaux 3b et 8c etant pratiquement identiques, la substitution des protons en tete de pont par des methyles ne change done pas la conformation de la molecule.
Le radical 3b Ctant trap peu soluble, nous avons seulement CtudiC le radical &. En general, le deplacement paramagn~tique d’un proton, ou dun groupe de protons equivalents, observe sur le spectre de RMN, est propo~ionnel’9 5 la constante de couplage de l’electron non apparie avec le ou les protons consider&. La mesure des d~p~dcements para~gn~tiques permet done de determiner la grandeur et le signe de ces couplages. Pour une temperature T = 3WK et une frtquence de 250 MHz, nous avons aH(G) = -5.39 x W’(S,,, - S,&, ou spa,* reprtsente le d&placement observe, mesure en Hz par rapport au tttramithylsilane (TMS) pris comme reference interne et compte le positivement vers tes champs forts, et oi 8~;~ represente le deplacement chimique par rapport au TMS qu’aurait le proton consider6 en l’absence d’interac-.
242 A. RASSAT et J. ROSZAL!D
tion avec l’klectron non apparik Ce dkplacement est estimk d’aprts les spectres de RMN des amines corres- pondantes.
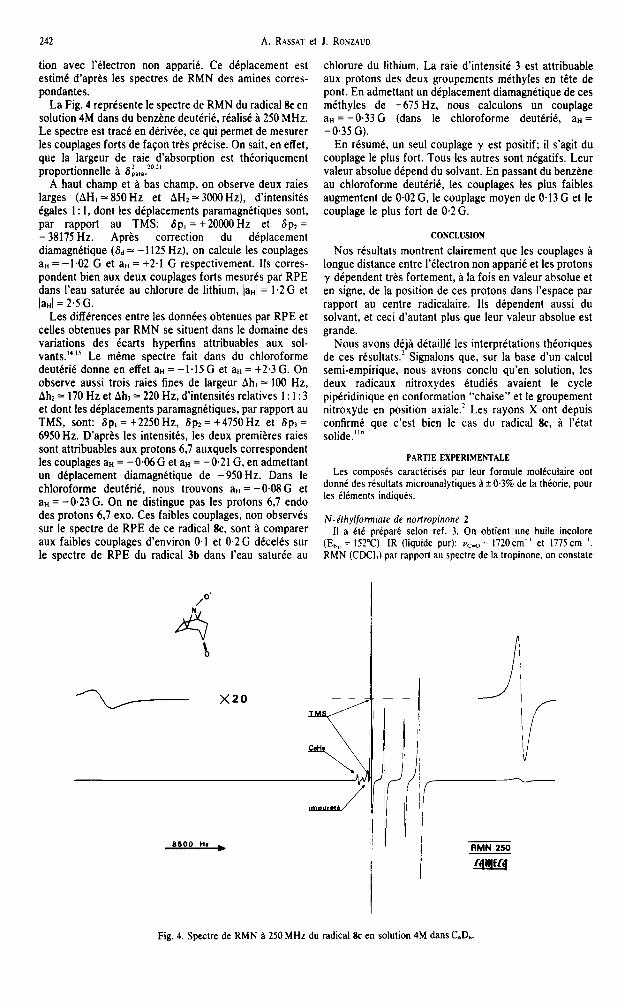
La Fig. 4 reprtsente le spectre de RMN du radical 8e en solution 4M dans du benztne deut%, rkalist g 250 MHz. Le spectre est track en dtkivte, ce qui permet de mesurer les couplages forts de faGon trks prkcise. On sait, en effet, que la largeur de raie d’absorption est thkoriquement proportionnelle B S i,,..“.*’
A haut champ et B bas champ, on observe deux raies larges (AH, = 850 Hz et AH2 = 3000 Hz), d’intensitks &ales 1: 1, dont les dkplacements paramagnktiques sont, par rapport au TMS: 6p, = +20OOOHz et Sp, = - 38175 Hz. Aprks correction du diplacement diamagnttique (& 3 - II25 Hz), on calcule les couplages aH = -1.02 G et aH = +2.l G respectivement. IIs corres- pondent bien aux deux couplages forts mesurks par RPE dans l’eau saturte au chlorure de lithium, JaHI = I *2 G et laHl = 2.5 G.
Les diffkrences entre les donntes obtenues par RPE et celles obtenues par RMN se situent dans le domaine des variations des &rts hyperfins attribuables aux sol- vants.“.” Le mCme spectre fait dans du chloroforme deutkrik donne en effet aH = -1.15 G et alI = t2.3 G. On observe aussi trois raies fines de largeur Ah, = 100 Hz, AhI = 170 Hz et Ah, = 220 Hz, d’intensitks relatives I : I : 3 et dont les dtplacements paramagnktiques, par rapport au TMS, sont: 6p, = +2250Hz, Sp, = +4750 Hz et Sp, = 6950 Hz. D’aprts les intensitks, les deux premiltres raies sont attribuables aux protons 6.7 auxquels correspondent les couplages aH = -O%G et aH = -0.21 G, en admettant un dkplacement diamagnktique de -950 Hz. Dans le chloroforme deuttrik, nous trouvons aH = -0.08 G et aH = -0.23 G. On ne distingue pas les protons 6,7 endo des protons 6,7 exo. Ces faibles couplages, non observCs sur le spectre de RPE de ce radical 8c, sont h comparer aux faibles couplages d’environ 0.1 et 0.2G d&elks sur le spectre de RPE du radical 3b dans I’eau saturke au
chlorure du lithium. La raie d’intensite 3 est attribuable aux protons des deux groupements mtthyles en tite de pont. En admettant un diplacement diamagnktique de ces mkthyles de -675 Hz, nous calculons un couplage aH = -0.33 G (dans le chloroforme deutCri& a” = -0.35 G).
En r&urn& un seul couplage y est positif; il s’agit du couplage le plus fort. Tous les autres sont nkgatifs. Leur valeur absolue depend du solvant. En passant du benztne au chloroforme deutkrik, les couplages les plus faibles augmentent de 0.02 G, le couplage moyen de 0.13 G et le couplage le plus fort de 0.2G.
CONCLUSION
Nos rksultats montrent clairement que les couplages g longue distance entre I’Clectron non apparik et les protons y dkpendent trks fortement, g la fois en valeur absolue et en signe, de la position de ces protons dans I’espace par rapport au centre radicalaire. IIs dtpendent aussi du solvant, et ceci d’autant plus que leur valeur absolue est grande.
Nous avons dkja dktaillk les interpktations thkoriques de ces rkultats. Signalons que, sur la base d’un calcul semi-empirique, nous avions conclu qu’en solution, les deux radicaux nitroxydes Ctudits avaient le cycle pipkridinique en conformation “chaise” et le groupement nitroxyde en position axiale.’ Les rayons X ont depuis confirm6 que c’est bien le cas du radical k, g 1’Ctat solide.“h
PARTIE EXPERIMENTALE
Les composk caract&& par leur formule mokulaire ant do& des rksultats microanalytiques B + 0.3% de la thkorie, pour les tlkments indiquk.
N-ithylfomiale de nortropinone 2
II a ktk prepare selon ref. 3. On obtient une huile incolore (E,,, = 152°C). IR (liquide pur): vc_,, = 1720cm- et 1775 cm ‘. RMN (CDCI,) par rapport au spectre de la tropinone, on constate
8500 HZ RMN 250
Fig. 4. Spectre de RMN g 250 MHz du radical lk en solution 4M dans C6D6.

Nitroxydes-LXX1 243
la disparition du methyle (2.45 ppm) et l’apparition du triplet (1.3 ppm) et du quadruplet (4.25 ppm) caracteristiques du group&
ment ethyle.
Nortropinone 3a On d&out I g de 2 darts 4 cm’ de diethylene glycol et 20ml
d’une solution 2N de soude. On laisse sous azote B 75°C pendant 6 h, avec forte agitation. Apres refroidjssement de la solution on passe en milieu acide (H,SO,). II se produit un degagement gazeux pendant environ l/2 h. La solution est lavee & Y&her ethylique. La phase aqueuse est neutralisie dans la glace par de la potasse en pastiltes, saturee au carbonate de potassium et extraite a l’tther en continu pendant 48 h. Apres sechage sur sulfate de sodium et evaporation du solvant sous vide, on obtient un liquide brun huileux que l’on chromato~phie sur colonne d’alumine basique activite V:2 fractions au pentane, puis 2 fractions a 2%, S%, 10%. 2.5%, 50% et 100% d’tther Cthylique. Enfin, en 2 fractions de 50% de chlorure de methylene, toute I’amine passe, soit 200 mg (3070 de rendement par rapport au N-Cthylformiate de nortropinone) que I’on peut encore recristalliser dans I.&her de p&role. Les cristaux blancs obtenus s’hydratent ou se carbonatent presque imm~diatement. IR (Nujol): Ye_” = 3350cm ’ et Ye.+,= 1775 cm-‘. RMN (CDCh): signal assez large d’intensite 2, sit& a 3.8 ppm, et caracteristique des deux protons en t&e de pont. Picrate (ethanol) C,,H,,O,N, (1, H. !$). F= 1909c (sur bane Kofler).
Un melange de 7 g d’hexane dione-2,s et dune solution de I7 g d’aeide acetone-dicarboxytique dans 100 ml d’eau est neutralisi dans la glace par de la potasse en pastilles. On y verse ensuite une solution de IO g de chlorure d’ammonium et de IO g da&ate de sodium dans 100 ml d’eau. Le pH du melange est port& a 9 par de la potasse en pastilles, puis est laisse a la temp~rat~e ambiante, sous agitation pendant 3 jours. On lave la solution brune en milieu acide (HSO,) par du chlorure de mbthylene. On porte la phase aqueuse en milieu basique (KOH), on la sature au carbonate de potassium et l’extrait B I’tther Bthylique en continu pendant 48 h. Apres sechage sur Na,SO, et ~va~ration sous vide du solvant, on obtient une huile bnme que I’on N&e sur une colonne d’alumine basique d’activite V, avec ilution a l&her de p&role. On obtient environ 2g dune huile Claire. On chromatographie cette huile sur colonne d’alumine basique, activite V. Apres Clution a l&her de p&role, puis Z+ p&her de p&role contenant 2%. S%, 18% et 20% d’ether ethylique, on obtient environ 1.5 g de cristaux jam&es que l’on recristallise dans du pentane distill& (20% de rendement par rapport B la dicetone de depart). Sous forme hydra&, l’amine forme des paillettes blanches, F = 44°C (sur bane Kofler). IR (Nujol): vy_,, = 1720cm ‘, “Y-H = 3350cm-‘, Y&- = 805, 1700, 2100 et 23OOcm“: voH = 368Ocm I. RMN (CDCI?): un pit d’intensite 6 B 1.35ppm attribue aux deux methyles en t&te de pont, un pit d’intensitt 4 a 1.7 ppm attribut apres deut~riation aux protons lies aux carbones 6 et 7, un pit d’intensitb 4 ti 2.3 ppm, attribue apres deuteriation aux protons lies aux carbones 2 et 4, un pit d’intensite variant entre 1 et 2 B 2.5 ppm attribud au proton de la fonction amine secondaire. Picrate (methanol), CIliH,*OsN, (c, H, N, Q), F = 22O’C (sur appareil Biichi).
On echauge dans l’eau lourde 7 g d’hexane dione-2,S en milieu basique. On obtient la didtone deutdriee a911 (spectrographic de masse). Un melange de 5 g d’hexane dione-2,5 deuteriee 191% et de 14g ~acide-acetone dic~boxylique dissous dans lOOmI d’eau lourde est neutralise par de la potasse en pastilles. On y verse ensuite une solution de 6 g de chlorure d’ammonium et de 6 g da&ate de sodium dans 100 ml d’eau lourde. Le pH du melange est port6 a 9 par de la potasse en pastilles. Le melange est agite et laisse a la temperature ambiante pendant 7 jours. L’extraction et la p~~~cation se font ensuite comme dans le cas de l’amine non deuteriee. Par rapport au spectre de RMN de la dimethyl-I,5 nortropinone, on constate la disparition quasi totale du pit 2 Me (I.35 ppm) et la disparition dun pie d’intensite 4 (I .‘l ppm) que l’on peut done attribuer aux protons lies aux carbones 6 et 7.
Nonropinone nitroxyde 3b On dissout 500 mg de 3a dans 4 cm’ d’eau. On ajoute 1 ml d’eau
oxygknte P 110 volumes et 40 mg d’acide phosphotun~tique. On laisse le melange B agitation pendant 2 h. Cette solution resee est saturee au chlorure de sodium. Le radical est extrait au chlorure de mithylene. Apres sechage sur sulfate de sodium et evaporation sous vide du solvant, on obtient environ 300 mg de produit brut radicalaire, sous forme de poudre jaune. Le radical est purifie par chromatog~pbie sur colonne ~alumjne neutre, activite V: la poudre est dissoute dans un peu de chlorure de mCthylene que l’on depose en haut de la colonne; apres tlution au pentane, puis au pentane contenant IO%, 20%, 48% et 6@% d’fther Qhylique, on obtient 15Omg de poudre jaune que I’on recristallise dans un melange 40% pentane -60% ether ethylique. On obtient ainsi environ 100 mg de radical cristallise (cristaux jaunes) F = 89aC (sur appareil Biichi). IR (Nujol): v+(,= I’flScm ‘. UV (chloroforme) e4,,, = 8.5, Ebb= 3000. RPE (solution 2 x IO-’ M dans I’eau saturee au chlorure de lithium): aN = 20.25G, a,,(ZH) = 5.75 G, a,,(2H) = 2.5 G, a”(2H) = I.25 G, a”(2H) -0.2 G et a”(2H) = 0.1 G. C7Hlo0,N (C, H, N).
~~m~t~yl-l,S no~ropi~one ~itroxyde 8e On dissout 500 mg de 8b dans 4 cm’ d’eau. On ajoute I ml d’eau
oxygenee h I IO volumes et SO mg d’acide phosphotungstique. On laisse le melange a agitation pendant 2 h et demie. Cette solution orangee est saturee au chlorure de sodium. Le radical est extrait au chlorure de methylene lavt trois fois par 100cmJ da&de sutfurique normal. Le solvant est s&he sur sulfate de sodium et evaport sous vide. On obtient 300 mg d’une huile rouge que l’on purifie par chromatographie sur 30 g d’alumine neutre, activite III. On tlue simplement a l’ether de p&role. Par recristallisation dans l&her de p&role refroidi B la carboglace, on obtient environ 200 mg de radical cristalfid. Les cristaux sont oranges. F = 44.YC fsur appareil B&hi). IR (Nujol): vcn = 1720 cm-‘. UV (cyclohe- xane)f eua = 10, c z45 = 2500. RPE (solution 2 x 10-3M da& l’eau saturee au chlorure de lithium): aK = 19.5 G, a”(2H) = 2.5G, an(2H) = 1.1 G. CsH,,OzN CC, H, N, 0).
‘A. Rassat et P. Rey, Tetrahedron 29.2845 (1973): et references intemes.
‘A. Rassat et J. Ronzaud, J. Am. C/tern. Sot. 93.5041 (1971); les mesures de RMN ont et& faites sur un spectre en absorption obtenu par un spectrom~tre Varian HA. 100 trav~llant dans des conditions non prevues par le constructeur et, de ce fait, beaucoup moins sensible et p&is que le spectrombtre Cameca que nous utilisons ici.
‘B. J. Calvert et J. D. Hobson, J. Chem. Sot. 2723 (1965). T. A. Montzka, J. D. Matiskella et R. A. Partyka, Tetrahedron Letters 1325 (1974): G. Kraiss et K. Nador. Ibid. 57 (1971).
‘R. J. Bishop, G. Fddor, A. R. Katritzky, F. Soti, L. E. &tton et F. J. Swin~urne, J. Chem. Sot. (Cj 74 (19%) et references intemes: H. Schenk. C. H. McGillavrv. S. Skolnik et J. Laan. Acta Cryst. 23, 423 (l%?). *
*K. Kovacs, K. Koczka. K. Thuransky, P. Agocs et I. Weisz, Brevet Hongrois 154574 (30 septembre 1968).
‘L. A. Paquette et J. W. Heimastek, J. Am. Chem. Sot. 88,763 (1966); eirhfkrences intemes.
‘R. Briere. H. Lemaire et A. Rassat, Bull. Sot. Chim. Fr. 3273 (1%5); G: Chapelet-Letoumeux, H. Lemaire et A. Rassat, Ibid 3283 (1%5).
‘R. M. Dupeyre et A. Rassat, J. Am. Chem. Sot. 88,318O ( 1%). “‘.A. Capiomont, B. Chion et J. Lajzerowicz, Acfo Crysi. 27B, 322
(1971); hA. Capiomont, Ibid. 29B, 1720 (1973). ‘I4 F. Genoud, M. C. Schouler et M. Decorps, Chem. Bps. Letters
26, 414 (1974); bM. Decorps et COB., travail en tours. “G. D. Mendenhall et K. U. Ingold, 1. Am. Chem. Sot. 9s. 6390 (1973); R. M. Dupeyre. A. Ra&at et J. Ronzaud, a publier.
“C. Vevret et A. Blaise, Not. Phvs. 28. 873 (1973). “R. Sri&e, H. Lemaire et A. Rassat, ~etr~h~d~~~ Letters 1775
ww. “R. Briere, A. Rassa?, P. Rey et B. Tchoubar, .i. C&n. Phys. 1575
(1966).

244 A. RASSAT et J. RONZAU~
“E. G. Rozantsev et D. Cholle, Dokl. Akad. Nauk. SSSR 187, “K. H. Hausser, H. Brunner et J. C. Jochims, Mol. Phys. 10,253 1319 (1969). (I%@.
“A. F. Thomas, Deuterium Labeling in Organic Chemistry, pp. “R. Briere, H. Lemaire. A. Rassat, P. Rey et A. Rousseau, Bull. 181-185. Meredith, New York (1971). Sot. Chim. Fr. 4479 (1%7).
“A. Rassat et J. Ronzaud, en tours de publication. “R. W. Kreilick. Mol. Phgs. 14, 495 (1966).





























