Embed Size (px)
Citation preview

SYNTHÈSE D’UN CYCLOPROPANE VINYLIQUE
ET ÉTUDE DE SON COMPORTEMENT D’OUVERTURE
par
Patrick Cyr
Sous la supervision de Martin Déry
Rapport de stage T0
Présenté au Professeur Claude Spino
FACULTÉ DES SCIENCES
UNIVERSITÉ DE SHERBROOKE
Sherbrooke, Québec, Canada, août 2010

ii
REMERCIEMENTS
J’aimerais remercier le professeur Claude Spino pour m’avoir accepté dans son laboratoire pour mon
stage T0. Ensuite, j’aimerais remercier mon superviseur Martin Déry pour son aide, son appui et sa
patience, ainsi que tous les autres membres du laboratoire : Dana Winter, Francis Beaumier, Jasmin
Douville, Simon Pichette, Pascal Léveillé, Louis-Philippe D. Lefebvre et Marie-Michèle Cournoyer.
J’aimerais aussi remercier tous les autres membres du département de chimie de l’université de
Sherbrooke pour avoir transformé ce stage T0 en une belle expérience enrichissante.

iii
TABLE DES MATIÈRES
REMERCIEMENTS ................................................................................................................................. II
TABLE DES MATIÈRES ....................................................................................................................... III
LISTE DES TABLEAUX ........................................................................................................................ IV
LISTE DES SCHÉMAS ............................................................................................................................ V
INTRODUCTION ...................................................................................................................................... 1
I.1 Antécédents littéraires ................................................................................................................. 1
I.2 Présentation du projet .................................................................................................................. 4
CHAPITRE 1 : SYNTHÈSE DU CYCLOPROPANE VINYLIQUE ....................................................... 5
CHAPITRE 2 : EXPLORATION DE LA RÉACTIVITÉ DU CYCLOPROPANE .................................. 9
2.1. Introduction ................................................................................................................................ 9
2.2. Hypothèses, résultats et discussions .......................................................................................... 9
2.2.1. Réactions connues de la littérature sur des cyclopropanes vinyliques .......................... 9
2.2.2. Réactions moins présentes dans la littérature des cyclopropanes vinyliques ............. 12
CONCLUSION GÉNÉRALE .................................................................................................................. 17
PARTIE EXPÉRIMENTALE .................................................................................................................. 18
Remarques générales ...................................................................................................................... 18
Modes opératoires ........................................................................................................................... 20
RÉFÉRENCES ET NOTES ..................................................................................................................... 24
ANNEXE 1 : SPECTRES DE RÉSONANCE MAGNÉTIQUE NUCLÉAIRE DES PROTONS .......... 25

iv
LISTE DES TABLEAUX
Tableau 1 : Première étape de la synthèse de départ. ....................................................................................... 6
Tableau 2 : Modifications au réarrangement de Johnson‐Claisen. ..................................................................... 6

v
LISTE DES SCHÉMAS
Schéma 1 : Exemple d’ouverture avec l’étain ................................................................................................... 1
Schéma 5 : Synthèse du cyclopropane ............................................................................................................. 5
Schéma 6 : Prédiction du radical formé durant le mécanisme .......................................................................... 9
Schéma 7 : Réaction radicalaire prévue ......................................................................................................... 10
Schéma 8a : Réaction radicalaire observée .................................................................................................... 10
Schéma 8b : Diagramme d’énergie ................................................................................................................ 11
Schéma 9 : 5+2 intermoléculaire .................................................................................................................... 12
Schéma 10 : Réaction radicalaire A avec le bromobenzène ............................................................................ 13
Schéma 11 : Réaction radicalaire avec le bromobenzène, deuxième essai ...................................................... 13
Schéma 12 : Réaction radicalaire avec un allylstannane ................................................................................. 14
Schéma 13 : Réaction de bromation avec NBS ............................................................................................... 15
Schéma 14 : Réaction de bromation avec Br2 ................................................................................................. 15

1
INTRODUCTION
Récemment, le groupe de recherche du professeur Claude Spino a fait des études
sur des réactions de cycloaddition 4 + 1 entre des carbènes riches en électrons et des diènes pauvres en
électrons. Ils ont pu déterminer que différents produits étaient obtenus dépendamment de la pauvreté en
électron du diène. En effet, ils ont montré qu’attacher un groupement électroattracteur au diène favorise
la conception de la cyclisation par la formation du produit de cycloaddition [4+1], tandis que l’absence
d’un tel groupement sur ce même diène provoque plutôt une cyclopropanation. Les carbènes utilisés
sont des dialkoxycarbènes, ce qui leur confère leur caractère riche en électrons. Donc, lorsque l’on
provoque une cyclopropanation, on obtient un cyclopropane vinylique avec deux alkoxy rattachés sur
lui. Ce genre de molécules est plutôt nouveau dans la littérature. C’est pourquoi l’étude de la réactivité
de ce dernier serait très didactique et pourrait engendrer une utilisation particulière dans la synthèse de
molécules naturelles.
I.1 Antécédents littéraires
Pour pouvoir se lancer dans un tel projet, il a été primordial de se renseigner sur
l’avancement de la connaissance dans ce domaine très spécialisé de la chimie organique. Plusieurs
types de réactions ont déjà été étudiés sur les cyclopropanes. Par exemple, la littérature démontre la
possibilité de passer par une ouverture par voie radicalaire du cyclopropane comme on peut le voir dans
une publication de Rahm et Degueil-Castaing (voir schéma 1)1.
4 ̊C 76% 24% 55 ̊C 61% 39%
Schéma 1 : Exemple d’ouverture avec l’étain
Cette réaction est très révélatrice dans notre cas, puisqu’elle démontre la possibilité
d’ouvrir le cyclopropane. À noter qu’ils font réagir leur système sous haute pression d’azote (14 kbars).
HSnMe3
Me3Sn
+ SnMe3
1 23
Température Rendements
14 kbars

2
De plus, fait intéressant, ils réussissent à contrôler le pourcentage de produit final dépendamment de la
température. Plus la température était élevée, plus le produit 3 était obtenu en grande proportion. C’est
donc qu’une température élevée favorise l’ouverture du cyclopropane. Malheureusement, les
rendements de l’ouverture du cyclopropane demeurent plutôt faibles et il sera donc important d’essayer
de trouver les conditions dans lesquelles ils seront agrandis avec le moins de produits secondaires
possibles. Il serait également intéressant de pouvoir prédire le côté d’ouverture du cyclopropane en
partant d’une molécule chirale.
C’est pourquoi le travail de Katsukiyo Miura et son équipe devient très intéressant
pour nous.2 Ils ont démontré la réaction du schéma 2 qui illustre une façon radicalaire d’ouvrir un
cyclopropane.
Schéma 2 : Exemple d’ouverture par voie radicalaire
Cette réaction suscite de l’intérêt puisqu’elle révèle une belle possibilité
d’ouverture d’un cyclopropane. Elle nous démontre que les cyclopropanes s’ouvrent à l’aide de
réactions radicalaires avec l’hydrure de tributylétain. On constate aussi que l’ouverture du cyclopropane
se fait par le lien unique qui relie le vinyle au reste de la molécule. Ceci est expliqué par la stabilité
accrue du radical, celui-ci étant tertiaire et stabilisé par résonance avec les deux esters, formé après
l’addition du radical stanane dans le mécanisme. De plus, ils obtenaient de très bons rendements pour
cette réaction, ce qui est de bon augure. En résumé, il est donc évident que l’ouverture du cyclopropane
est possible de façon radicalaire.
O
O
O
OAIBN
HSnBu3Bu3Sn
O
O
O O
4 5

3
Ensuite, on peut aussi trouver dans la littérature des exemples de réactions [5+2]
inter ou intra moléculaire avec des cyclopropanes. Le schéma 3 est un bel exemple de [5+2] intra
moléculaire avec un catalyseur de rhodium publié par Stephen F. Martin 3 et le schéma 4 un exemple de
[5+2] inter moléculaire, toujours avec le rhodium, publié par Paul A. Wender.4
Schéma 3 : Exemple de [5+2] intra moléculaire
Schéma 4 : Exemple de [5+2] inter moléculaire
De plus, on peut remarquer que pour la version intramoléculaire, l’ouverture du
cyclopropane se fait assez bien avec un rendement de 85%. De son côté, avec des conditions
légèrement différentes, l’ouverture par voie inter moléculaire est aussi très bonne avec un rendement de
93%. Par ailleurs, en créant un bicycle, cette réaction est aussi intéressante du point de vue de la
synthèse de molécules naturelles, où on retrouve régulièrement ce genre de squelette. L’ouverture du
cyclopropane par [5+2] semble donc une belle approche qui a de solides antécédents littéraires.
OHC
MeO2C
MeO2C
OBn
H
H
[Rh(CO)2Cl]2, PhMe
110 °C, 85%
MeO2C
MeO2CH
OBn
CHO
6 7
O
O
OMe
[RhCl(cod)]2, AgSbF6
DCE, t.a.93%
OMeO
8 9

4
I.2 Présentation du projet
Ce projet de recherche, concernant principalement la chimie d’un cyclopropane
vinylique, a pour but premier l’exploration de la réactivité de ce dernier. Plusieurs publications ont déjà
apporté plusieurs réponses comme vu plus haut, mais certaines interrogations demeurent quant à la
réactivité de notre système plutôt particulier. En effet, le cyclopropane visé lors de ce projet est rattaché
par deux alkoxys, ce qui rend les réactions vu précédemment incertaines. Ce projet a donc pour but de
synthétiser ce dit cyclopropane et de tester sa réactivité en explorant différentes réactions possibles.
Premièrement, il faudra tester les réactions déjà connues dans la littérature du
même type que montré plus haut pour pouvoir assurer qu’elles peuvent être viables avec notre
molécule. On parle ici d’ouverture par voie radicalaire avec l’étain et d’ouverture selon une réaction de
[5+2]. À noter que la réaction du schéma 1 est aussi intéressante, mais demande des montages à haute
pression. Deuxièmement, il est possible de pousser la recherche vers des réactions moins connues dans
la littérature. Toutes sortes de possibilités s’ouvrent quant à l’exploration de la réactivité du
cyclopropane, autant ionique que radicalaire. Il sera permis de vérifier si faire réagir un électrophile
avec la double liaison vinylique au cyclopropane peut faire ouvrir le cyclopropane. Ensuite, il serait
possible de faire réagir la molécule avec des sulfures ou des halogénures qui pourraient de la même
façon, possiblement venir additionner sur la double liaison en ouvrant le cyclopropane. De plus, une
simple réaction comme l’ajout d’une chaîne alkyle ou d’un phényle sur un allylstannane de façon
radicalaire pourrait théoriquement dégager l’étain en additionnant sur la double liaison liée au
cyclopropane. C’est donc avec ces objectifs que la réactivité du cyclopropane sera explorée.
5
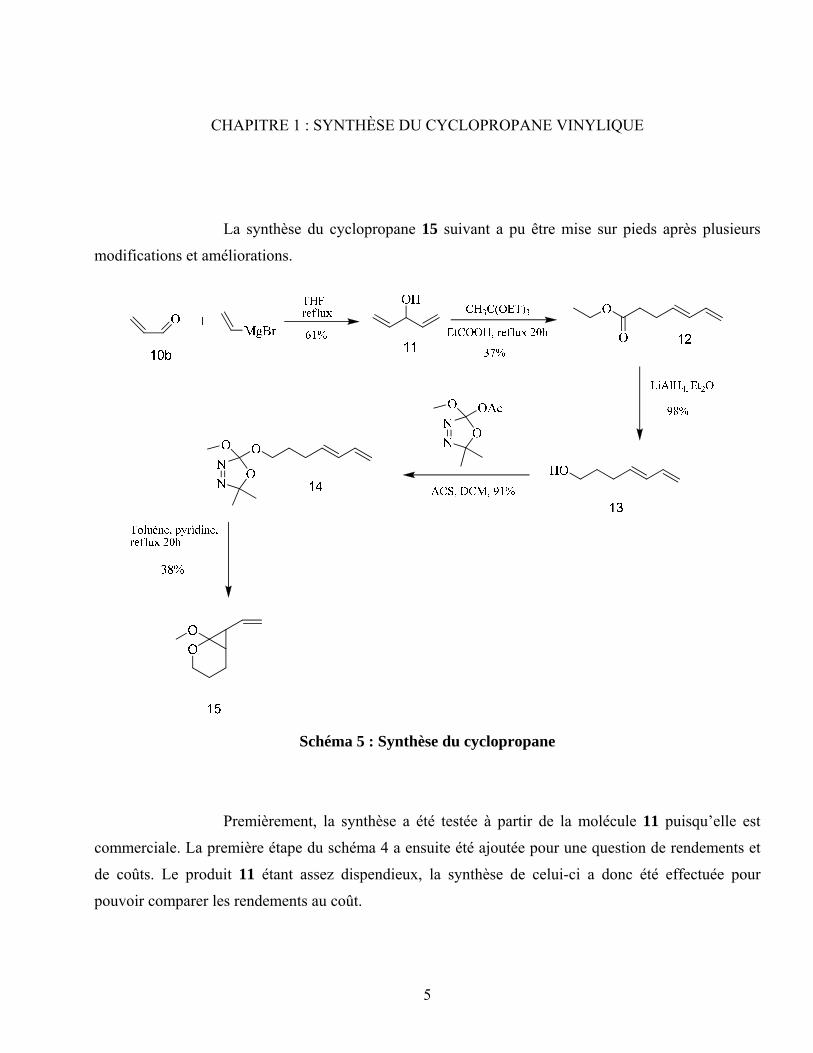
CHAPITRE 1 : SYNTHÈSE DU CYCLOPROPANE VINYLIQUE
La synthèse du cyclopropane 15 suivant a pu être mise sur pieds après plusieurs
modifications et améliorations.
Schéma 5 : Synthèse du cyclopropane
Premièrement, la synthèse a été testée à partir de la molécule 11 puisqu’elle est
commerciale. La première étape du schéma 4 a ensuite été ajoutée pour une question de rendements et
de coûts. Le produit 11 étant assez dispendieux, la synthèse de celui-ci a donc été effectuée pour
pouvoir comparer les rendements au coût.
6
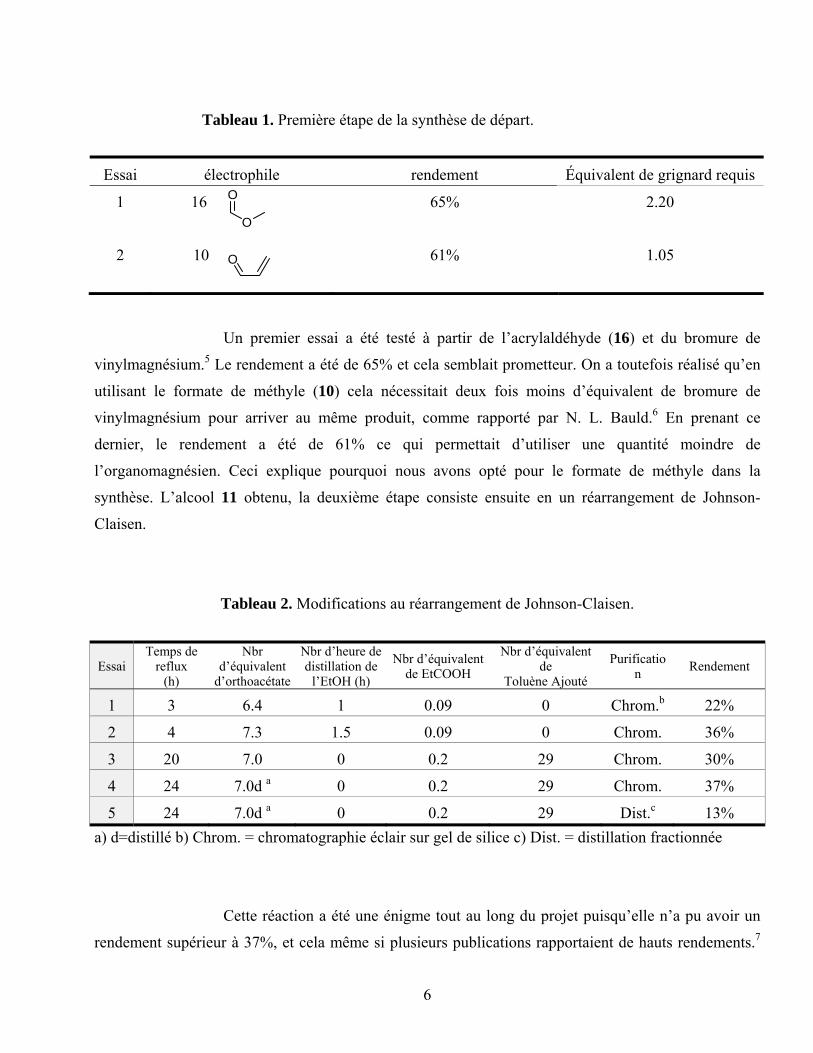
Tableau 1. Première étape de la synthèse de départ.
Essai électrophile rendement Équivalent de grignard requis
1 16
65% 2.20
2 10
61% 1.05
Un premier essai a été testé à partir de l’acrylaldéhyde (16) et du bromure de
vinylmagnésium.5 Le rendement a été de 65% et cela semblait prometteur. On a toutefois réalisé qu’en
utilisant le formate de méthyle (10) cela nécessitait deux fois moins d’équivalent de bromure de
vinylmagnésium pour arriver au même produit, comme rapporté par N. L. Bauld.6 En prenant ce
dernier, le rendement a été de 61% ce qui permettait d’utiliser une quantité moindre de
l’organomagnésien. Ceci explique pourquoi nous avons opté pour le formate de méthyle dans la
synthèse. L’alcool 11 obtenu, la deuxième étape consiste ensuite en un réarrangement de Johnson-
Claisen.
Tableau 2. Modifications au réarrangement de Johnson-Claisen.
Essai Temps de
reflux (h)
Nbr d’équivalent
d’orthoacétate
Nbr d’heure de distillation de
l’EtOH (h)
Nbr d’équivalent de EtCOOH
Nbr d’équivalent de
Toluène Ajouté
Purification
Rendement
1 3 6.4 1 0.09 0 Chrom.b 22%
2 4 7.3 1.5 0.09 0 Chrom. 36%
3 20 7.0 0 0.2 29 Chrom. 30%
4 24 7.0d a 0 0.2 29 Chrom. 37%
5 24 7.0d a 0 0.2 29 Dist.c 13%
a) d=distillé b) Chrom. = chromatographie éclair sur gel de silice c) Dist. = distillation fractionnée
Cette réaction a été une énigme tout au long du projet puisqu’elle n’a pu avoir un
rendement supérieur à 37%, et cela même si plusieurs publications rapportaient de hauts rendements.7
O
O
O

7
Comme le tableau 2 le rapporte, la réaction a d’abord été tentée en distillant l’éthanol produit durant la
réaction. Après un rendement de 22%, la réaction a été réessayée en suivant par RMN1H l’éthanol
distillé. La solution recueillie s’est avérée être un mélange d’éthanol et de triéthyl orthoacétate. Cette
procédure a donc été abandonnée, d’autant plus qu’elle était beaucoup plus complexe. Ensuite, la
réaction a été testée sur un plus long temps de chauffage sans distillation de l’éthanol. Ne donnant
toujours pas un rendement satisfaisant, nous avons décidé de prendre un RMN1H du triéthyl
orthoacétate utilisé afin de vérifier sa pureté. Ce dernier s’est révélé être mélangé à de l’acétate d’éthyle
1:1 et à de l’éthanol 1 :1. Pour optimiser la réaction, le triéthyl orthoacétate a donc été distillé pour le
purifier. Cependant, la distillation ne l’a pas totalement rendu pur. On remarque pourtant une hausse
minime du rendement à 37%. Finalement, la réaction a été réessayée en purifiant le produit brut par
distillation fractionnée pour compenser la possible perte de rendement en raison de la légère volatilité
du produit final. Cependant, le produit obtenu était très visqueux et semblait avoir polymérisé et a dû
être purifié par chromatographie éclair pour donner un maigre rendement de 13%. Le temps manquant
pour optimiser cette réaction davantage, la procédure a été gardée comme étant celle ayant obtenu 37%
de rendement.
Ensuite, l’alcool 13 est obtenu par la réduction au LAH de l’ester 12. Après avoir
constaté des rendements plutôt bas en raison de l’addition du LAH à l’ester, nous avons ajouté l’ester
au LAH pour obtenir des rendements excellents et constants.8 Par la suite, il suffisait de substituer
l’acétate d’un oxadiazoline par l’alcool 13. L’oxadiazoline utilisé comme réactif avait déjà été préparé
en deux étapes par mon superviseur Martin Déry. Après quelques essais, il s’est avéré nécessaire de
mettre 2 équivalents d’oxadiazoline afin d’atteindre la complétion de la réaction. La seule difficulté
majeure de la réaction fut la purification du produit 14 obtenu. En effet, une espèce minoritaire se
formait constamment et a été identifiée comme étant de l’hexa-3,5-diènyl méthyl carbonate. La cause
de sa formation a été déterminée comme étant l’ouverture de l’oxadiazoline par des traces d’acide dans
le milieu. Ce dernier était très dur à séparer par chromatographie, il fallait donc être très méticuleux
quant au montage et à l’éluant choisi.
La cinquième et dernière étape de cette synthèse consiste en une cyclopropanation.
Le carbène formé à partir de la fragmentation de l’oxadiazoline 14 a ainsi effectué une cycloaddition
[2 + 1] et le cyclopropane 15 a pu être obtenu. Les rendements étant assez bas pour cette réaction, nous
avons essayé de l’optimiser. Premièrement, la purification était aussi difficile qu’à l’étape précédente

8
en raison du même carbonate, provenant ici de la dégradation de l’oxadiazoline. C’est pourquoi un
traitement au méthanol et au carbonate de potassium a été testé afin de libérer le substrat sous forme
d’alcool qui est beaucoup plus polaire et plus facilement séparable par chromatographie éclair. Cette
technique s’est malheureusement soldée par un échec puisque le traitement n’a pas fait réagir le dit
carbonate. Deuxièmement, comme le mécanisme passe par la formation d’un carbène, il est essentiel de
rester dans des conditions anhydres. Malgré toutes les précautions prises pour enlever toutes traces
d’humidité, le rendement est demeuré stagné à 38% en étant tout de même constant.

9
CHAPITRE 2 : EXPLORATION DE LA RÉACTIVITÉ DU CYCLOPROPANE
2.1. Introduction
Le cyclopropane étant maintenant obtenu après la synthèse vu au chapitre un, il a
été possible de tester sa réactivité selon plusieurs réactions. Le but de cette démarche est, rappelons le,
de découvrir le comportement du cyclopropane soumis à différentes conditions puisqu’il est plutôt
singulier en ayant deux alkoxys rattachés à lui. Plusieurs hypothèses ont été testées tant au niveau de
réactions radicalaires qu’ioniques. Cependant, il a été plus sécuritaire de commencer par des réactions
déjà connues de la littérature.
2.2. Hypothèses, résultats et discussions
2.2.1. Réactions connues de la littérature sur des cyclopropanes vinyliques
Il a été avantageux de commencer l’exploration par une réaction connue de la
littérature qui aurait de grande chance de réussite. C’est pourquoi la décision a été prise de commencer
par une addition radicalaire par l’étain qui ouvrirait le cyclopropane (molécule 15). Comme présenté
dans l’introduction, ce genre de réaction est connu pour ouvrir du côté qui sera favorisé par la formation
d’un radical plus stable lors du mécanisme.
Schéma 6 : Prédiction du radical formé durant le mécanisme
O SnBu3O
OSnBu3
O
15 17

10
Le schéma 6 présente la partie décisive prévue du mécanisme, tout de suite après
qu’AIBN soit venu prendre l’hydrogène de l’hydrure de tributylétain. Comme vu dans la publication de
Katsukiyo Miura dans l’introduction2, la formation de radicaux est toujours favorisée par la stabilité de
ces derniers. Ici, la prédiction du schéma 7 a été influencée par deux aspects principaux. Premièrement,
en ouvrant de ce côté du cyclopropane, le radical formé est tertiaire, plutôt que secondaire. Comme le
démontre la littérature et la théorie, le radical tertiaire est plus stable que le secondaire. Deuxièmement,
les radicaux sont connus pour être plus stables s’ils sont stabilisés par des groupements
électrodonneurs. C’est donc que le radical hypothétiquement formé dans le schéma 6 est plus stable en
raison des deux oxygènes en alpha qui le stabiliseront par résonance.
Schéma 7 : Réaction radicalaire prévue
C’est avec un grand étonnement que nous avons constaté que la molécule 18
prévue n’était pas formée dans ces conditions. En effet, après avoir tenté de purifier le produit obtenu
par chromatographie éclaire, rien n’a été recueilli. Cependant, un autre essai a été tenté en suivant la
réaction par RMN1H en faisant celle-ci dans le benzène deutéré. Ceci a permis de découvrir qu’il y
avait bel et bien une ouverture du cyclopropane, mais pas selon nos prédictions. C’est donc que le
produit formé n’était pas assez stable pour résister sur la silice.
Schéma 8a : Réaction radicalaire observée
Le schéma 8a représente la vraie structure du produit retrouvée à la suite de la
réaction. Le mécanisme de la réaction passe donc par la formation du radical le moins stable. La raison
O
O
O
O
SnBu3
n-Bu3SnH, AIBN
Benzène, reflux15 18
O
O n-Bu3SnH, AIBN
Benzène, reflux O
OSnBu3
15 19
11
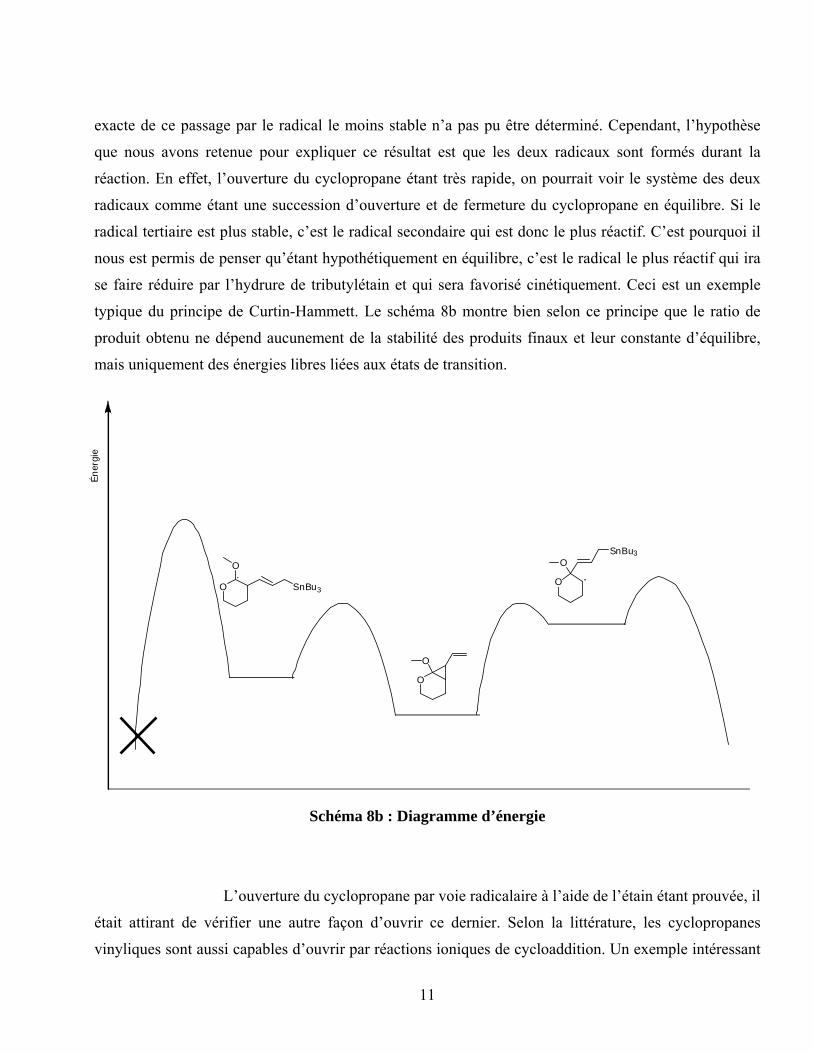
exacte de ce passage par le radical le moins stable n’a pas pu être déterminé. Cependant, l’hypothèse
que nous avons retenue pour expliquer ce résultat est que les deux radicaux sont formés durant la
réaction. En effet, l’ouverture du cyclopropane étant très rapide, on pourrait voir le système des deux
radicaux comme étant une succession d’ouverture et de fermeture du cyclopropane en équilibre. Si le
radical tertiaire est plus stable, c’est le radical secondaire qui est donc le plus réactif. C’est pourquoi il
nous est permis de penser qu’étant hypothétiquement en équilibre, c’est le radical le plus réactif qui ira
se faire réduire par l’hydrure de tributylétain et qui sera favorisé cinétiquement. Ceci est un exemple
typique du principe de Curtin-Hammett. Le schéma 8b montre bien selon ce principe que le ratio de
produit obtenu ne dépend aucunement de la stabilité des produits finaux et leur constante d’équilibre,
mais uniquement des énergies libres liées aux états de transition.
Schéma 8b : Diagramme d’énergie
L’ouverture du cyclopropane par voie radicalaire à l’aide de l’étain étant prouvée, il
était attirant de vérifier une autre façon d’ouvrir ce dernier. Selon la littérature, les cyclopropanes
vinyliques sont aussi capables d’ouvrir par réactions ioniques de cycloaddition. Un exemple intéressant
O
O
O
SnBu3
O
O
OSnBu3
Én
ergi
e

12
est les réactions de 5+2. Cependant, la plupart des publications traitent de 5+2 intramoléculaires comme
vu dans l’introduction. N’ayant pas la possibilité d’effectuer la réaction de manière intramoléculaire sur
notre système, nous avons cherché des références sur la possibilité de la faire de façon intermoléculaire.
Le travail de Paul A. Wender publié en 2010 sur les 5+2 intermoléculaires nous a servis de base solide
pour pouvoir tester notre cyclopropane 15.3
Schéma 9 : 5+2 intermoléculaire
Selon leur publication, le catalyseur et l’additif choisis ici (schéma 8) ne sont pas
les meilleurs. L’explication vient du fait que la réaction a été testée avec les produits déjà disponibles
au laboratoire en raison du manque de temps et du coût très élevé des autres options. Le suivi RMN1H
après 5h ne révélait plus de cyclopropane de départ. Le cyclopropane a définitivement réagi, mais suite
à une purification du produit obtenu, la molécule obtenue n’a pas pu être identifiée en raison d’un
manque de temps. Cependant, ce résultat est assez satisfaisant pour pouvoir poursuivre éventuellement
les études sur ce genre de réactions.
2.2.2. Réactions moins présentes dans la littérature des cyclopropanes vinyliques
Plusieurs autres réactions semblaient intéressantes à essayer sur notre système pour
bien maîtriser la réactivité du cyclopropane. Cependant, plusieurs de ces réactions n’ont guère
d’antécédents littéraires sur des systèmes comme le nôtre. Pour favoriser l’exploration, il a été décidé
de commencer par des réactions radicalaires puisque le cyclopropane ouvrait sans problème et assez
rapidement dans les conditions du schéma 8a.
O
O
RhCl(PPh)3, AgBF4
DCE/TFE 90:10, 60°C
O
O
O15
20
O

13
Schéma 10 : Réaction radicalaire A avec le bromobenzène
Cette réaction (schéma 10) était une tentative de chaîne radicale. Celle-ci consistait
à l’arrachement du proton du tributylétain pour former le radical d’étain. Ensuite, l’étain devrait
pouvoir venir chercher le brome du bromobenzène pour former un radical sur le benzène. Puis, le
radical du benzène viendrait additionner sur la double liaison qui entraînerait l’ouverture du
cyclopropane. Cette réaction a été testée en ajoutant l’hydrure de tributylétain et AIBN dans le benzène
sur une période de 2 heures à une solution contenant le cyclopropane et le bromobenzène dans le
benzène. Cette très lente addition (compte tenu du fait que la réaction se faisait sur 50 mg de
cyclopropane) a été réalisée pour avoir une plus grande quantité de substrat comparativement à
l’hydrure de SnBu3, ce qui améliore les chances d’attaquer le substrat. Malgré cette précaution, un suivi
par RMN1H a démontré que le cyclopropane n’a jamais réagi. Cependant il est assez difficile de
déterminer avec exactitude la cause de l’échec de la réaction.
Par la suite, loin d’abandonner du côté des réactions radicalaires, une réaction
similaire a été essayée à partir de la molécule 15. Ici, le but était surtout d’obtenir un produit ayant un
autre substituant que l’étain. Substituant qui serait plus stable et qui offrirait d’autres options de
réactions.
Schéma 11 : Réaction radicalaire avec le bromobenzène, deuxième essai
O
O
n-Bu3SnH, AIBN
Ph-Br, benzène 80°C
O
OPh
15 21
n-Bu3SnH, AIBN
Ph-Br, benzène 80°C
O
OSnBu3
19
O
O
22
Ph

14
La chaîne de radicaux n’ayant pas fonctionnée à partir du cyclopropane, elle a été
essayée sur la molécule 19 pour pouvoir essayer de comprendre à quel niveau la réaction du schéma 10
était déficiente. Le seul changement majeur de la procédure a été de ne mettre qu’une quantité
catalytique d’hydrure de tributylétain. De cette façon, il nous sera possible de vérifier si à la suite de
l’attaque sur l’allylstanane par le radical du benzène, un autre radical d’étain sera formé pour pouvoir
revenir chercher le brome du bromobenzène. Ainsi, cela créerait une boucle de réaction jusqu’à ne plus
avoir de produit de départ. L’étain étant en quantité catalytique, cela empêche la réduction directe du
radical benzène. Malheureusement, après avoir suivi la réaction par RMN1H, aucun produit 19 n’a été
observé et tout le cyclopropane de départ est resté intacte. Ceci pourrait être expliqué par le fait que le
phényle qui doit s’additionner sur la double liaison est incapable de le faire en raison de
l’encombrement stérique dû au carbone quaternaire en alpha de cette double liaison qui est assez
encombrant.
Pour palier à ce problème d’encombrement nous avons décidé de tester la réaction
sur un autre réactif que le bromobenzène.
Schéma 12 : Réaction radicalaire avec un allylstannane
Cette réaction était très intéressante puisque le mécanisme passe par la formation
du même radical que lors de la formation du produit d’étain 19. Ce radical devrait alors additionner sur
la double liaison de l’allylstannane pour ensuite rejeter un autre radical d’étain pour poursuivre la
réaction. C’est donc une autre réaction où l’hydrure de tributylétain est en quantité catalytique.
Cependant, comme le montre le schéma 12, la réaction fut un autre échec. Fait étrange remarqué lors
des suivis par RMN1H, la molécule 19 n’est pas produite du tout : le cyclopropane reste intacte. Une
explication possible est que l’allylstannane est plus favorisé pour être attaqué par l’étain. De plus, après
deux essais infructueux, nous doutons qu’un radical d’étain est réellement relâché à la suite d’une
O
O
15n-Bu3SnH, AIBN
benzène 80°C
SnBu3
O
OSnBu3
23

15
addition sur une double liaison, allylique à ce dernier, dans ces conditions. C’est pourquoi la décision a
été prise de laisser de côté les réactions radicalaires.
Par la suite, sachant que le cyclopropane ouvrait de façon radicalaire dans des
conditions standards, il était intéressant de voir comment il réagirait de manière ionique. L’idée la plus
simple était de faire réagir le cyclopropane avec un électrophile connu.
Schéma 13 : Réaction de bromation avec NBS
C’est sur le NBS que notre choix s’est arrêté. Il a été basé sur le fait qu’il y a
beaucoup d’antécédents dans la littérature de conditions de réactions le concernant. Cependant, la
question était de savoir si le NBS serait assez puissant pour venir additionner sur la double liaison
vinylique au cyclopropane. Malheureusement, après plusieurs heures de reflux, le suivi par RMN1H a
clairement démontré que les produits de départ étaient restés intacts dans la solution.
Il fallait donc un électrophile plus puissant que le NBS pour faire réagir notre
molécule. Nous avons décidé de réessayer une réaction de bromation, mais cette fois à l’aide de Br2. Le
brome étant plus réactif, nous espérions pouvoir ouvrir le cyclopropane à l’aide de ce dernier.
Schéma 14 : Réaction de bromation avec Br2
O
O
15
O
O
Br
24
NBS
1:1 CH2Cl2/benzène
reflux
O
O
15
O
O
BrBr2
1:1 CH2Cl2/benzène
reflux 25

16
La réaction du schéma 14 a d’ailleurs fonctionnée. En effet, malgré un maigre
rendement de 24%, la molécule 25 a été identifiée par RMN1H et par spectre de masse. Nonobstant la
nécessité d’améliorer la procédure, la réaction est intéressante puisqu’elle prouve que le cyclopropane a
été ouvert par la simple addition d’un électrophile sur la double liaison. Il est aussi nécessaire de
mentionner que le produit obtenu n’a pu être assez purifié pour enlever toutes les impuretés.

17
CONCLUSION GÉNÉRALE
Étant donné que le projet principal était de synthétiser le cyclopropane 15 et
d’explorer sa réactivité, les résultats obtenus sont satisfaisants. Il a été permis de prouver l’ouverture du
cyclopropane par voie radicalaire et par voie ionique. Cependant, vu le manque de temps, plusieurs
réactions n’ont pas pu être testées. C’est pourquoi quelques projets futurs pourraient être intéressants à
explorer. Premièrement, il serait intéressant d’essayer d’identifier la molécule obtenue durant l’essai de
5+2. De plus, il serait utile d’utiliser un meilleur catalyseur. Un essai intra moléculaire serait aussi très
intéressant à observer. Un approfondissement des réactions radicalaires et leur optimisation serait aussi
une belle option pour continuer ce projet puisqu’il n’est pas parvenu à tirer profit de ces réactions. Plus
globalement, il serait très intéressant de trouver des voies de réactions possibles pouvant mener à
l’obtention de molécules naturelles à partir de ce type de système.

18
PARTIE EXPÉRIMENTALE
Remarques générales
Toutes les réactions ont été effectuées sous atmosphère d'argon dans de la verrerie
séchée à la flamme sous pression réduite. Les solvants anhydres et certains réactifs liquides ont été
distillés avant leur utilisation, et ils sont rapportés dans le tableau G.1 suivant.
Tableau G.1 : Agents desséchants utilisés pour la distillation de différents solvants et réactifs.
Solvant / Réactif distillé Agent desséchant
Benzène LAH
Dichloroéthane Hydrure de calcium
Dichlorométhane Hydrure de calcium
Éther diéthylique Sodium, Benzophénone
Pyridine Hydrure de calcium
Tétrahydrofurane Potassium, Benzophénone
Toluène Hydrure de calcium
Les chromatographies sur couche mince ont été effectuées sur des plaques de verre
recouvertes de gel de silice (0.25 mm, Silicyle). Les produits en chromatographie sur couche mince ont
été révélés à la lampe UV, puis par trempage dans une solution aqueuse de KMnO4 ou dans une
solution de vaniline, suivi d'un chauffage. Les chromatographies éclairs ont été effectuées avec du gel
de silice (40-63 m, Silicyle).

19
Les spectres infrarouges ont été obtenus par dépôt d'un film de produit sur une
pastille de bromure de potassium, avec un spectromètre Perkin-Elmer 1600 FT-IR. Les spectres de
résonance magnétique nucléaire (1H, 13C, DEPT) ont été enregistrés avec un appareil Bruker AC-300.
L’étalon interne est le chloroforme (7,26 ppm) pour la résonance des protons et le chloroforme (77,0
ppm) pour la résonance des carbones. Les spectres de masse ont été enregistrés avec un spectromètre
VG Micromass ZAB-2F.

20
Modes opératoires
(14) (E)-2-(hepta-4,6-dienyloxy)-2-methoxy-5,5-dimethyl-2,5-dihydro-1,3,4-oxadiazole
D’une solution de l’alcool PC-1-72-1 (0.914 g, 8.15 mmol) et de l’acide camphorsulfonique (0.11 g,
0.41 mmol) dans 47mL de dichlorométhane anhydre a été ajoutée une solution de l’oxadiazoline md1-
151-1 65% m/m (4.72 g, 16.3 mmol) dans 47 mL de dichlorométhane anhydre. La réaction a été agitée
pendant 1h à température ambiante. Une solution saturée de bicarbonate de sodium dans l’eau (90 mL)
a été ajoutée et les deux phases ont été séparées. La phase aqueuse a été extraite deux fois avec 150 mL
de dichlorométhane. Les phases organiques ont été combinées ainsi que séchées avec du sulfate de
magnésium anhydre pour ensuite être filtrées et évaporées sous pression à l’évaporateur rotatif. Le
produit brut recueilli a été purifié par une chromatographie éclair sur gel de silice utilisant comme
éluant un mélange d’acétate d’éthyle et d’hexane (5:95). Une huile incolore a été obtenue (1.79 g,
91%). RMN’H (300 MHz, CDCl3) (ppm) 6.30 (ddd, 1H, J = 17.0, 9.9, 9.9 Hz) 6.05 (dd, 1H, J =
15.4, 9.9 Hz) 5.68 (dt, 1H, J = 15.4, 6.6 Hz) 5.09 (d, 1H, J = 17.0 Hz) 4.97 (d, 1H, J = 9.9 Hz) 3.80-
3.62 (m, 2H) 3.46 (s, 3H) 2.17 (q, 2H, J = 7.1 Hz) 1.73 (dt, 2H, J = 7.1, 6.6 Hz) 1.55 (s, 3H) 1.56 (s,
3H). RMN 13C (75.5 MHz, CDCl3) (ppm) 137.0 (d), 137.0 (s), 133.8 (d), 131.6 (d), 118.8 (s), 115.0
(t), 63.9 (t), 51.7 (q), 28.9 (t), 28.7 (t), 24.0 (q), 24.0 (q). IR (CHCl3) ν (cm-1) 3085, 2989, 2948, 2846,
2365,2343, 1458, 1211, 1139. SMBR (m/z, intensité relative) 240 (M+, 65), 198 (55), 184 (50), 129
(55), 91 (75), 81 (100). SMHR calculée pour C12H20N2O3: 240.1474, trouvée: 240.1474.
N
NO
O O

21
(15) 1-methoxy-7-vinyl-2-oxabicyclo[4.1.0]heptane
Dans une verrerie préalablement lavée (ordre de lavage : hexane, dichlorométhane, acétoine, eau, acide
chloridrique conc., eau, hydroxyde de sodium conc., eau, éthanol ), de la pyridine (53 μL, 0.67 mmol)
est ajouté à l’oxadiazoline PC-1-85-1 (647 mg, 2.69 mmol) dans le toluène anhydre (solution 0.01 M).
Le mélange est chauffé à reflux pendant 20h. Le produit brut recueilli a été purifié par une
chromatographie éclair sur gel de silice utilisant comme éluant un mélange d’éther diéthylique et
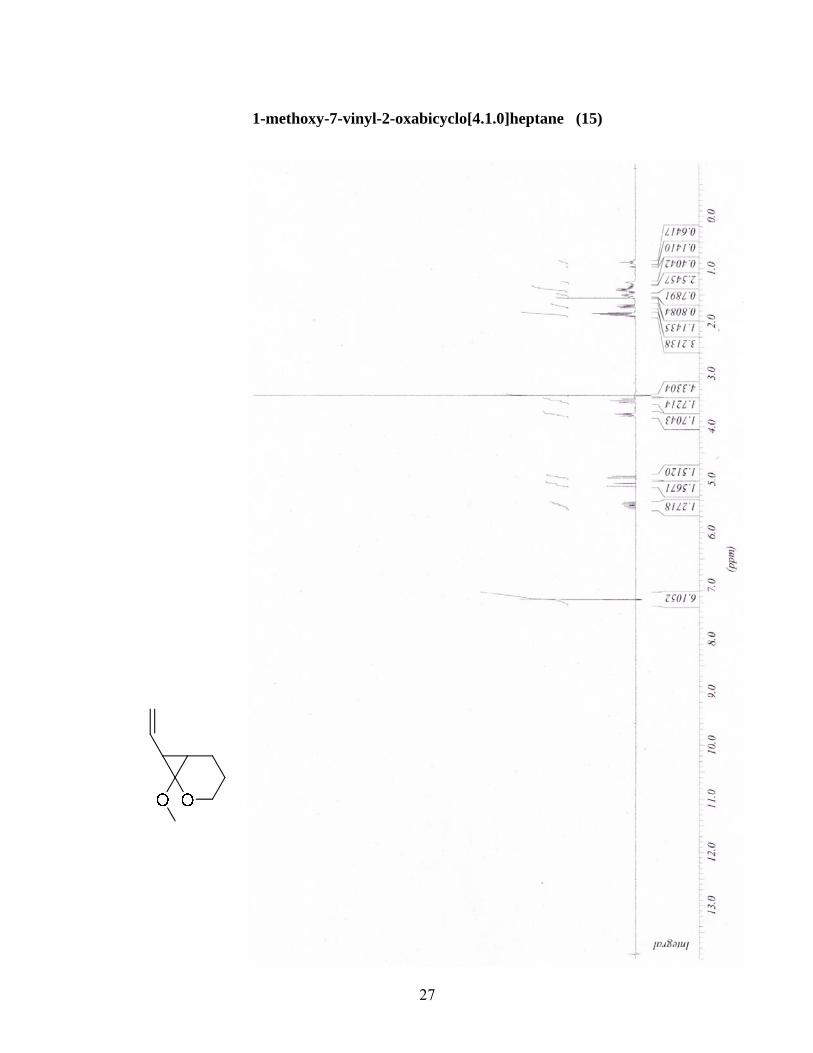
d’hexane (4:96 - 6:94 - 8:92). Une huile incolore a été obtenue (0.1576 g, 38%). RMN’H (300 MHz,
CDCl3) (ppm) 5.49 (ddd, 1H, J = 17.2, 10.1, 9.7 Hz) 5.10 (dd, 1H, J = 17.2, 1.6 Hz) 4.96 (dd, 1H, J
= 10.1, 1.6 Hz) 3.78 (dt, 1H, J = 10.4, 3.3 Hz) 3,51 (t, 1H, J = 10.4 Hz) 3.41 (s, 3H) 1.90-1.84 (m, 2H)
1.71 (dd, 1H, J = 9.7, 6.6 Hz) 1.55-1.50 (m, 1H) 1.42-1.38 (m, 2H). RMN 13C (75.5 MHz, CDCl3)
(ppm) 136.2 (d), 113.6 (t), 90.8 (s), 65.3 (t), 53.8 (q), 34.8 (d), 26.5 (d), 21.6 (t), 19.3 (t). IR (CHCl3) ν
(cm-1) 2949, 1736, 1440, 1253, 1054. SMBR (m/z, intensité relative) 154 (M+, 25), 142 (40), 114 (85),
97 (75), 83 (100). SMHR calculée pour C9H14O2: 154.0994, trouvée: 154.0993.
(19) (E)-tributyl(3-(2-methoxytetrahydro-2H-pyran-2-yl)allyl)stannane
O
O

22
Un mélange de PC-1-110-1 (56 mg, 0.36 mmol), d’hydrure de tertbutylétain (115 μL, 0.433 mmol) et
d’AIBN (12 mg, 0.072 mmol) a été chauffé à reflux dans le benzène anhydre (solution 0.02 M) pendant
1h en suivant la réaction par RMN1H. Le produit obtenu n’a pu être purifié et aucun rendement n’a pu
être observé. RMN’H (300 MHz, CDCl3) (ppm) 5.90 (dt, 1H, J = 15.4, 8.8 Hz) 5.11 (d, 1H, J = 15.4
Hz) 3.63 (dd, 2H, J = 8.5, 3.0 Hz) 3.10 (s, 3H) 1.74-1.69 (m, 4H) 1.51-1.44 (m, 8H) 1.30-1.23(m, 8H)
0.96-0.81(m, 15H). RMN 13C (75.5 MHz, CDCl3) (ppm) 132.0 (d), 126.2 (d), 97.8 (s), 61.1 (t), 48.3
(q), 36.0 (t), 29.0 (t), 27.6 (t), 25.0 (t), 19.1 (t), 14.1 (t), 13.6 (q), 9.9 (t). IR (CHCl3) ν (cm-1) 2955,
2925, 2866, 1460, 1228, 1155. SMBR (m/z, intensité relative) 389 (M-C4H9+, 6), 357 (23), 291 (70),
235 (75), 179 (95), 124 (100). SMHR calculée pour C21H42O2Sn – C4H9: 389.1502, trouvée: 389.1510.
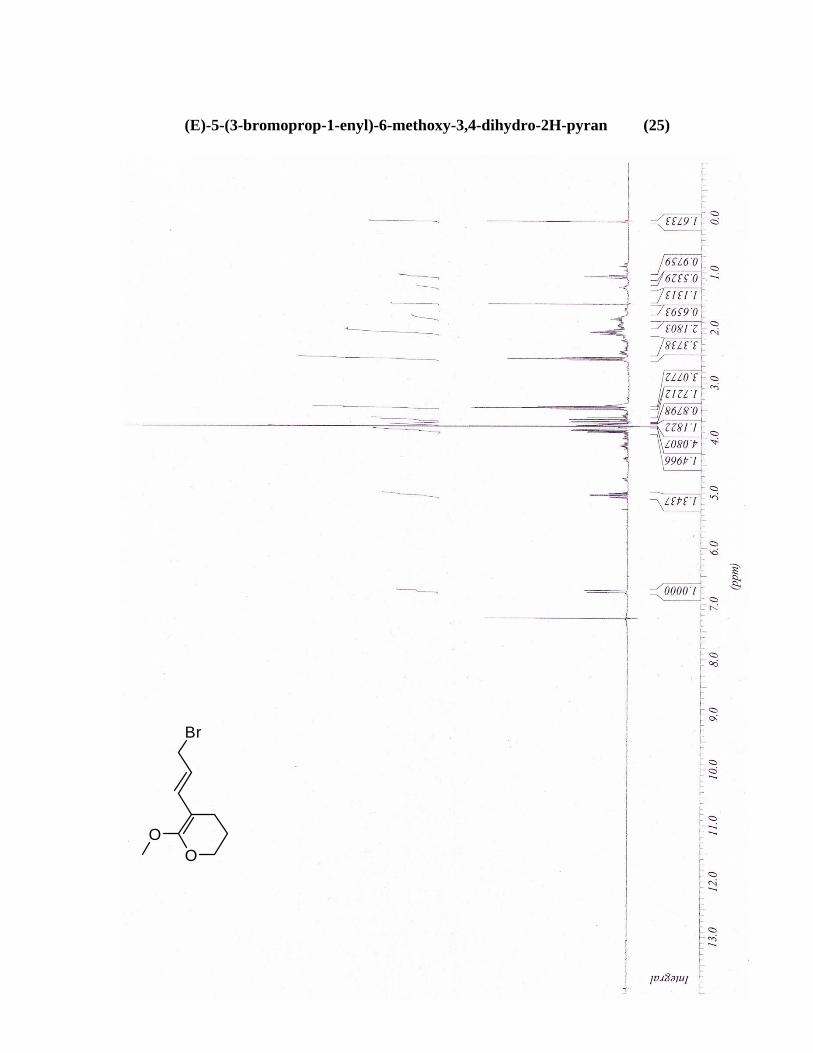
(25) (E)-5-(3-bromoprop-1-enyl)-6-methoxy-3,4-dihydro-2H-pyran
O
OSnBu3

23
Une solution de PC-1-110-1 (45 mg, 0.29 mmol) et Br2 (15 μL, 0.29 mmol) dans du benzène/DCM 1 :1
(3 mL) a été agitée à t.a. pendant 2h. Une solution d’acide acétique 1 N a été ajoutée (5 mL) et les deux
phases ont été séparées. La phase aqueuse est extraite 2 fois au DCM (2 x 5 mL). Les phases organiques
sont combinées et séchées avec du MgSO4 anhydre pour ensuite être filtrées et évaporées sous pression
à l’évaporateur rotatif. Le produit brut recueilli a été purifié par une chromatographie éclair sur gel de
silice utilisant comme éluant un mélange d’acétate d’éthyle et d’hexane (2:98). Une huile incolore a été
obtenue (18.1 mg, 24%). RMN’H (300 MHz, CDCl3) (ppm) 6.78 (d, 1H, J = 11.0 Hz) 5.05 (td, 1H, J
= 11.0, 4.4 Hz) 3.86 (d, 2H, J = 4.4 Hz) 3.79 (s, 3H) 3.45 (t, 2H, J = 6.6 Hz) 2.56 (t, 2H, J = 7.7 Hz)
2.11-2.03 (m, 2H). SMBR (m/z, intensité relative) 234 ((M+2)+, 10), 232 (M+, 10), 125 (70), 93 (100),
41 (30).

24
RÉFÉRENCES ET NOTES
1 Rahm,A.; Degueil-Castaing, M.; Pereyre, M. Journal of Organometallic Chemistry.1982,vol.
232, C29-C32 2 Miura, Katsukiyo; Fujisawa, Naoki; Saito, Hiroshi; Nishikori, Hisashi; Hosomi, Akira. Chem.
Let., 2002, 32-33. 3 Brandon, L. Ashfeld; Stephen, F. Martin. Organic Letters, Vol.7, No.20, 2005, 4535-4537. 4 Paul A. Wender; Lauren E. Sirois; Rene T. Stemmler and Travis J. Williams. Organic Letters,
Vol.12, No.7, 2010, 1604-1607. 5 Hugh, E. Ramsden; Jack, R. Leebrick; Sanders, D. Rosenberg; Edith, H. Miller; John, J. Walburn;
Allen, E. Balt and Robert, Cserr. Journal of organic chemistry, Vol. 22, 1602-1605 6 N. L., Bauld. Journal of physical organic chemistry, Vol.11, 1998, 825-830 7 Lynn, C. Baillie ; Andrei, Batsanov ; John, R. Bearder and Donald, A. Whiting. J. Chem. Soc.
Perkin Trans., Vol. 1, 1998, 3471-3478
Claude, Spino; Jason, Crawford and John, Bishop. Journal of organic chemistry, 1995, 60, 844-
851 8 E., Vedejs ; T. H., Eberlein and R. G., Wilde. Journal of organic chemistry, 1988, 53, 2220-2226

25
ANNEXE 1 : SPECTRES DE RÉSONANCE MAGNÉTIQUE NUCLÉAIRE DES PROTONS
26
(E)-2-(hepta-4,6-dienyloxy)-2-methoxy-5,5-dimethyl-2,5-dihydro-1,3,4-oxadiazole (14)
27
1-methoxy-7-vinyl-2-oxabicyclo[4.1.0]heptane (15)

28
(E)-tributyl(3-(2-methoxytetrahydro-2H-pyran-2-yl)allyl)stannane oxadiazole (19)
29
(E)-5-(3-bromoprop-1-enyl)-6-methoxy-3,4-dihydro-2H-pyran (25)
OO
Br

























![Chrom journal8 fr[1]](https://img.pdfslide.fr/doc/110x75/55700931d8b42ac0178b4776/chrom-journal8-fr1.jpg)

