Embed Size (px)
Citation preview

1
1
Pathologies Hémorragiques
Institut de formation Infirmier en Anesthésie réanimation
Dr Pierre Fialon
Laboratoire de Biologie LRRCHU de Bordeaux – Hôpital Saint André
Juillet 20122
Les pathologies hémorragiques congénitales
� relativement rares. � déficit quantitatif ou qualitative d’un seulfacteur de la coagulation ou d’un seul constituant plaquettaire.
� Les hémorragies surviennent � dés la naissance au moment de la coupure du cordon ombilical.
� +tardivement dans l’enfance voire à l’age adulte dans les formes frustes
� au moment d’un acte opératoire d’ou l’importance de l’examen clinique et biologique pré opératoire.
3
Pathologies hémorragiques acquises
� très fréquentes� déficit combinés de plusieurs facteurs de la coagulation
� Les hémorragies surviennent dans l’évolution de plusieurs pathologies et peuvent être révélatrice.
� Exceptionnellement elles surviennent dés la naissance : carence en vitamine K
4
Examen d’un trouble
hémorragique � Contexte:
� pré opératoire
� manifestations hémorragiques
� Objectifs� Confirmer la symptomatologie ou le risque
� Apprécier la gravité
� Examen clinique (exhaustif si hémorragies)
� Interrogatoire (check liste)
� Antécédents personnels et /ou familiaux
� Traitements en cours ou récents
� Bilan biologique (nombreuses formes frustes)
5
Pathologies congénitales
6
Pathologies de l’hémostase primaire
ThrombopéniesThrombopathies
Maladie de Willebrand
2
7
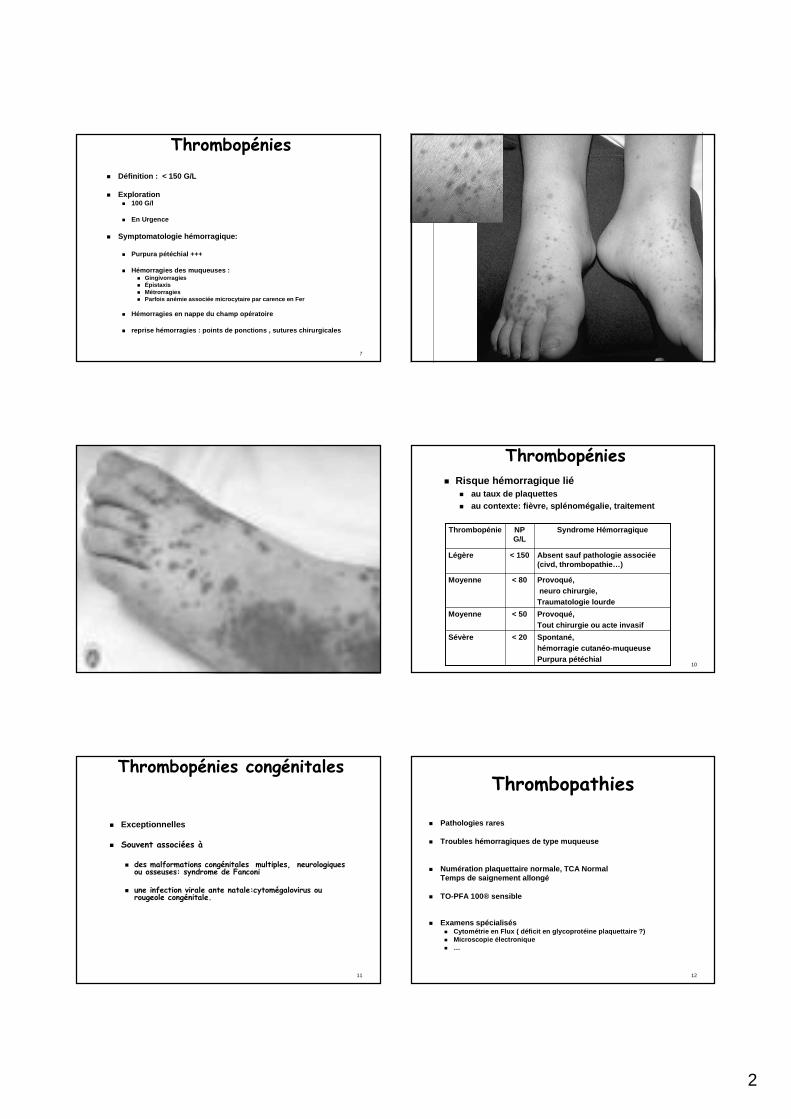
Thrombopénies� Définition : < 150 G/L
� Exploration� 100 G/l
� En Urgence
� Symptomatologie hémorragique:
� Purpura pétéchial +++
� Hémorragies des muqueuses : � Gingivorragies � Épistaxis � Métrorragies� Parfois anémie associée microcytaire par carence en Fer
� Hémorragies en nappe du champ opératoire
� reprise hémorragies : points de ponctions , sutures chirurgicales
8
9 10
Thrombopénies� Risque hémorragique lié
� au taux de plaquettes� au contexte: fièvre, splénomégalie, traitement
Provoqué,neuro chirurgie, Traumatologie lourde
< 80Moyenne
Spontané, hémorragie cutanéo-muqueusePurpura pétéchial
< 20Sévère
Provoqué, Tout chirurgie ou acte invasif
< 50Moyenne
Absent sauf pathologie associée (civd, thrombopathie…)
< 150Légère
Syndrome HémorragiqueNP G/L
Thrombopénie
11
Thrombopénies congénitales
� Exceptionnelles
� Souvent associées à
� des malformations congénitales multiples, neurologiques ou osseuses: syndrome de Fanconi
� une infection virale ante natale:cytomégalovirus ou rougeole congénitale.
12
Thrombopathies
� Pathologies rares
� Troubles hémorragiques de type muqueuse
� Numération plaquettaire normale, TCA NormalTemps de saignement allongé
� TO-PFA 100® sensible
� Examens spécialisés� Cytométrie en Flux ( déficit en glycoprotéine plaquett aire ?)� Microscopie électronique� …

3
13
THROMBOPATHIES
� Maladie de Bernard et Soulier� Absence glycoprotéine plaquettaire GP Ib� Transmission autosomale récessive
� Thrombasthénie de Glanzmann� Absence GP IIb IIIa� Transmission autosomale récessive
� Maladie du pool vide� Anomalie stockage (ADP) / sécrétion granules denses
� Syndrome de Scott� Flipflop anormal des phospholipides membranaires
� Syndrome des plaquettes grises� Anomalie stockage des protéines (granules alpha)
/ 60 14
THROMBOPATHIES: traitements
� Limitation exposition aux risques hémorragiques
� Utilisation de traitements locaux:� les hémorragies externes ++++
� Les actes médico chirurgicaux locaux : chirugiedentaire, ORL, cutanée, …
� Les concentrés de plaquettes d’aphérèse (CPA)� les accidents graves,
� la chirurgie ou les accouchements
� Minirin ® desmopressine
/ 60
15
Maladie de Willebrand
16
MALADIES DE WILLEBRAND� Anomalie de l'hémostase la plus fréquente
� Secondaire à une anomalie quantitative ou qualitative du facteur VIII Willebrand
� Transmission autosomale dominante : touche les deux sexes.
� Les manifestations cliniques débutent dès l’enfance:� hémorragies des muqueuses : épistaxis, melæna, ménorragies et ecchymoses.
� L’aspirine aggrave la symptomatologie.
� Formes modérées sans accidents hémorragiques spontanés� dépistées lors d’un bilan de coagulation systématique
� Rares formes très sévères homozygotes avec des hémarthroses.
� Le diagnostic biologique est évoqué devant :� temps de saignement allongé
� taux de plaquettes normal
� TP normal
� TCA allongé
� Confirmation est obtenue par un dosage du facteur Willebrand.
17
MALADIES DE WILLEBRAND
� Explorations complémentaires: type et sous-type� Répartition des multimères, vWF Ag, vWF RCo dans les
plaquettes,RIPA� Étude liaison vWF-F VIII, Étude liaison vWF-Plaquettes, � Analyse ADN
� Classification� TYPE 1 : anomalie quantitative
� le plus fréquent
� TYPE 2 : anomalies qualitatives diverses,� individualisation de différents sous types (2A, 2B, 2N, 2M…)� 2B thrombopénie (desmopressine non utilisable)
� TYPE 3 : absence totale de facteur Willebrand� clinique voisine de celle de l'hémophilie
18
Principaux type de MW
x
X
N
↓
<0,7
↓
N
N
X
N
↑
x
Anomalie liaison GP
Ib
2 M
X1<0,7<0,71RCo / Ag
↓ ↓ ↓↓ ↓N ou ↓N ou ↓N ou ↓F VIII c
↑ ↑ ↑N↑↑+/- ↑TS
↑ ↑ ↑↑+/- ↑+/- ↑+/- ↑TCA
NN↓NNNum P
TotalDéfaut liaison vWF / VIII
Augmentation affinité vWF /
GP Ib/IX
Défaut liaison vWF/Plaquettes
PartielDéficit vWF
Récess.Récess.Dom.Dom.Dom.Transmission
Concentrés de vWF
Concentrés de vWF
Concentrés de vWF
Concentrés de vWF
DDAVPTraitement
1-3%?3-5%10-12%70-80%Fréquence
AbsentsNAbs HPMAbs IPM et HPM↓ (N)Multimères
AbsenteAbsenteAugmentéeAbsenteAbsenteRIPA
AbsentN↓ ou ↓ ↓↓ ↓ ↓↓vWFRCo
AbsentN+/- ↓↓↓vWFAg
32N2B2A1Type
! TCA Normal n’exclut pas MW

4
19
MALADIES DE WILLEBRAND: traitement
� Prise en charge par un centre de référence: CRTHCentre Régional de Traitement des maladies Hémorrag iques
� Information et éducation des patients, importance d es traitement mineurs eviter acte hémorragique inutile
� Traitement par desmopressine MINiRIN ®� Hormone antidiurétique
� Sécrétion de facteur Willebrand de la cellule endothéliale
� Augmente par 2 ou 3 taux de facteur
� Phénomène d’épuisement de l’effet thérapeutique� Effets indésirables fréquents (> 1/100, < 1/10) : céphalées, douleurs abdominales,
nausées, congestion nasale
� Accident rare intoxication par l'eau :vomissements, anorexie, augmentation rapide du poids corporel, état confusionnel, convulsions dans les cas sévères, hyponatrémie et hypo-osmolalité
arrêt traitement
� Test au Minirin préalable avant chirurgie programmée
20
MALADIES DE WILLEBRAND: traitement
� Substitution par Médicaments Dérivés du Sang� Fractionnement du plasma� Synthétisé après recombinaison génétique
� Composition� Facteur Willebrand Pur� Facteur Wilebrand associé à Facteur WIIIC anti Hémophi lique
� Protocole : CRTH ou médecin référents du patient
� Traitement à démarrer 8 heures avant ou au moment de l’intervention et à perfuser soit àla seringue électrique soit en perfusion discontinu e (2 à 3 par Jour)
� Gestion rigoureuse des thérapeutiques� Traçabilité� Conservation� Cout
� Surveillance clinique et biologique (TCA, Facteur Wi ilebrand)Hémostase assurée lorsque:
facteur VIII coagulant (FVIII:C) > 0,4 Ul/ml (40 %)
facteur VWF willebrand > 0,6 Ul/ml (60%)
21
Subtitution
injections
WIII vwF
100 %
60 %
20 %
40 %
80 %
Temps en jours
1 43 52
22
Pathologies de la coagulation
Hémophilies A & BDéficit I, II,V,VII,,X, XI,XII
23
Hémophilie A & B
24
Hémophilie� Maladie hémorragipare héréditaire grave
� hémophilie A = déficit en FVIII 1/5000 naissance � (2500 hémophiles en France)
� hémophilie B = déficit en FIX 1/30000
� Récessif lié à X L’affection ne touche que les garçons
Les filles indemnes de manifestations peuvent trans mettre l’anomalie.
� Sévérité fonction taux de facteurs VIII ou IX� Sévère < 2 %
� Modérée 2 - 5 %
� Mineure 5 - 40 %

5
25
Hémophilie� Clinique
� Débute tôt dans l’enfance : apprentissage de la mar che
� Hémarthroses; genou, cheville, du coude et de la hanche�
� hématomes,
� hémorragies post-traumatiques ou post chirurgicales en l’absence de traitement
� Hémorragie viscérale : rares ( facteur déclenchant surajouté ?)
� Fréquence variable� Hémophilie sévère: plusieurs fois par mois, traumat isme souvent inaperçu
� Hémophilie modérée: rarement et en cas de choc impo rtant
� Hémophilie mineure: uniquement per opératoire ou po st traumatique
26
Hémophilie� Risques liés aux localisations particulières
Certaines localisations nécessitent un traitement important et souvent l’hospitalisation du patient afin d’éviter des complications par compression
� Hématomes cervicaux et du plancher de la bouche pouvant être responsables d’asphyxie.
� Hématomes des membres qui sont sources de compression vasculaire et nerveuse avec risque d’installation d’un syndrome de Volkmann lorsque le siège est au niveau de l’avant bras ou un équinisme du pied pour les hématomes du mollet
� Hématome du psoas ou de la paroi intestinale qui simule une urgence chirurgicale abdominale: appendicite, occlusion
� Hématomes rétro-orbitaires : risque de cécité.
27
Hémophilie� Complications
� Risque: développement d’un inhibiteur neutralisant les traitements substitutifs
� Anti VIII (20% des patients)
� Anti IX (3%)
� Transmission d’agents infectieux
� Arthropathie chronique
Séquelle des hémorragies intra articulaire avec déformation ou blocage articulaire
responsable d’un handicap fonctionnel majeur.
28
Hémophilie: diagnostic biologique
� Interrogatoire� Atcd hémorragie, hématomes� Arbre généalogique...
� Augmentation TCA avec TP et TS normaux
� Dosage VIII (A) & IX (B)
� Recherche anti VIII & anti IX� Apparition / thérapeutique substitutive� Recherche régulière et avant tout acte chirurgical
� Dépistage ante natal: génomique ou fonctionnel
29
Hémophilie: traitement� SUBSTITUTIF CURATIF
� Facteur IX ou VIIIc peut être apporté par les fractions purifiées à partir du fractionnent du plasma humain ou par des médicaments préparés par génie génétiques.
� Incidents mineurs: doit être réalisé le plus rapidement possible, si possible à domicile, par le patient ou sa famille ; dans l’hémarthrose simple : une seule injection de 20 UI/kg
� Les patients doivent être en relation avec un centre régional de traitement de l’hémophilie.
� Pour les urgences il est impératif de contacter leur médecin hémophilologue et veiller à transfuser le médicament habituel.
� TRAITEMEMENT ADJUVANTS� produits hémostatiques locaux
� antalgiques dépourvus d’aspirine et d’anti-inflammatoires non stéroïdiens
� intra-musculaires sont à proscrire.30
Hémophilie: traitement
� SUBSTITUTIF Préventif
� Consiste en l’administration régulière de facteurs de coagulation pour prévenir l’apparition de saignements.
� Ces facteurs sont injectés aux patients deux à trois fois par semaine, ce qui permet de transformer une forme sévère de l’hémophilie en une forme modérée.
� Ce traitement améliore l’ état ostéo-articulaire. .

6
31
Déficits en fibrinogène,facteurs II, V, VII, X
� TCA allongé ( sauf facteur VII), TP allongéavec ou sans tendance hémorragique,
� Déficit complet en fibrinogène � Exceptionnel
� Traitement : apport fibrinogène
� Dysfibrinogénémie� Risque de thrombose
� Déficit II, VII, X, VHétérozygote Homozygote
� Rare Exceptionnel
� TP voisin de 50% TP < 15 %
� pas de risque hémorragique Risque hémorragique
� Pas de traitement Traitement
- Déficit II, VII, X : PPSB
- Déficit V : PFC 32
Allogngement isolé du TCA,facteurs XII, XI VIII, IX
XII
XI
IX
VIII
VII
X
II
I
V
TCA Allongé TQ Normal
Déficit en facteur ?
XII
XI
IX
VIII
Anticoagulant circulant ?:
héparine,
anticoagulant
33
Allogngement isolé du TCA,facteurs XII, XI VIII, IX, VII, X
� TCA allongé , TP Normal
� avec ou sans tendance hémorragique
� A explorer en urgence en pré opératoire
� Risque hémorragique
� Déficit XI, VIIIc (Hémophilie A) , VIII vWF(Willebrand), IX
� anticoagulant thérapeutique
� Risque thrombotique
� Déficit en facteur XII
� Anticoagulant circulant antibphospholipides
� Intérêt d’anticiper la réalisation des bilans pré opératoires34
Pathologies acquises
35
Pathologies de l’hémostase primaire
ThrombopéniesThrombopathies
36
Thrombopénies acquises � Fréquentes
� Exploration en urgence : ponction sternale� Pas de mégacaryocyte : mécanisme central� Mégacaryocyte nombreux : mécanisme périphérique
� Thrombopénies centrales:
� Étiologie� Atteinte toxique : irradiation, chimique ou pharmaceutique� Aplasie� Envahissement médullaire :métastase, leucémie, lymphome
� Traitement � Spécifique de l’étiologie� Transfusion de concentré de plaquettes si pronostic favorable

7
37
Thrombopénies acquises
� Thrombopénies périphériques :
� Consommation� CIVD� Hémangiomes géants� Circuit extra vasculaires : CEC� Micro angiopathie thrombotique
� Destruction immunologique� PTI Purpura thrombopénique immunologique
� Étiologies� Affections virales (infection banale enfant, VIH)� Maladie de système� Idiopathiques � Traitement: exemple : thrombopénie à l’héparine
� Traitement� Immunologique
� Corticoïde , immunoglobulines polyvalente intraveineuses� Immuno supresseur� Splénectomie
� Arrêt médicament (38
Thrombopathies
� Pathologies très fréquentes
� Aspirine chez enfant� Aspirine et anti-inflammatoire chez adulte � Anti agrégeant plaquettaires chez sujet agé
� Risques per opératoire � Troubles hémorragiques si non arret du traitement� Thromboses si arrêt anti agrégeant
� Attitude à envisager au cas pas cas en fonction de� Type anti agrégeant: durée action � Intervention� Risque thrombotique
� Exemple: arrêt aspirine 4 à 5 Jours avant chirurgie chez patient avec stent actif
39
Pathologies de la coagulation
Déficit en facteur vitamino K dépendantCIVD
Insuffisance hépato cellulaireAnticoagulant circulant
Iatrogènes40
Déficit en facteur vitamino K dépendant� Baisse du TP et un allongement du TCA� Baisse de facteurs II, VII et X alors que le facteur V et le fibrinogène sont normaux.
� Etiologies� Carence d’apport, patients dénutris soumis à une alimentation parentérale et à une désinfection intestinale.
� Trouble de l’absorption.- diarrhée chronique (maladie coeliaque)- résection intestinale étendue- ictère choléstatique (calcul du cholédoque, cancer de la tête du pancréas
� Carence d’utilisation induite par la prise d’anti-vitamine surdosage ++++
41
Déficit en facteur vitamino K dépendantTraitement
� Fonction de l’étiologie et de la gravité.
� vitamine K per os: efficace en 24 ou 48 heures
� intra musculaire : réservé à traitement préventif
� injection intra -veineuse lente (30Mn): en 6 à 12 heures
� Fraction PPSB � 10ml/10 kg de poids correction immédiate
� utilisation déconseillée si insuffisance hépatique
42
Coagulation Intra VasculaireDisséminée
CIVD

8
43
Coagulation Intra VasculaireDisséminée
� Urgences thérapeutiques.
� activation exagérée de la coagulation qui va entraîner la formation de micro thromboses puis secondairement l’apparition d’un syndrome hémorragique et d’une fibrinolyse réactionnelle.
� Les fibrinolyses pures sans C.I.V.D. sont plus rares. Elles sont responsables d’accidents hémorragiques
44
Activation exagérée de la coagulation
Facteurs Contacts
Facteur Tissulaire
PLCa2+VIIIa
PK, KHPM, XIIa, XIa, IXaVIIa
PLCa2+
X XaPLCa2+V
II IIa
Fibrinogène Fibrine
Voie endogène
Voie exogène
-AT 3
45
Coagulation Intra Vasculaire DisséminéeCirconstance étiologiques
� favorisée par l’existence d’une insuffisance hépato-cellulaire et les états de choc avec anoxie.
� déclenchée par le passage dans le sang de thromboplastines tissulaires :
� Les hémolyses intra-vasculaires aiguës: transfusion ABO incompatible
� Septicémie : méningite à méningocoque� Les polytraumatismes et les grandes interventions chirurgicales
� Leucémies LAM 3 � Chirurgie ou les cancers du poumon, de la prostate� Complications obstétricales : embolie amniotique, fœtus mort, toxémie gravidique,Hématome rétroplacentaire
� Venin de serpents 46
Coagulation Intra Vasculaire DisséminéeTableau clinique
� Manifestations thrombotiques : � Troubles neurologiques (coma)� Lésions cutanées (purpura nécrotique en carte de géographie, gangrène)
� Lésions rénales (oligurie, anurie par insuffisance rénale fonctionnelle puis irréversible)
� Troubles pulmonaires (insuffisance respiratoire aiguë)
� Manifestations hémorragiques� Hémorragie en nappe au niveau du champ opératoire� Hémorragie incoercible aux points de prélèvements et aux sutures des plans opératoires
� Purpura ecchymotique extensif en carte de géographie
47 48
CIVD: DIAGNOSTIC BIOLOGIQUE� Numération plaquettaire: Diminution
� Allongement des tests globaux de la coagulation� TCA, TP, TT
� Diminution des facteurs de la coagulation� Fibrinogène ���� ���� ����
� Facteurs � V,VIII, XII, X, XII, prékallicréine ���� ���� ����
� VII, IX, X + stables � II variable � V plus bas que II, VII et X
� Signes d’hyper-fibrinolyse� PDF
� Produits de dégradation de la fibrine et du fibrinogè ne� D-Dimères
� Générés par l’action de la plasmine sur la fibrine

9
49
Plasmine Plasminogène
PDF: Produit de dégradation de la fibrine
D Dimère fragment de 2 chaînes
Avtivateur du Plasminogène
Caillot de fibrine
Hyper coagulation
50
COAGULATIONThrombine
Fibrinogène Caillot deFibrine
PDFplasmatiques
PDF D Dimères
Plasmatiques
Plasmine PlasmineFIBRINOLYSE FIBRINOLYSEprimitive réactionelle
51
CIVD: Traitement� Moyens thérapeutiques
� Traitement de l’étiologie et correction de l’état de choc.
� Traitement substitutif : Transfusion de plaquettes, de PFC.
� Transfusion de concentré d’anti-thrombine III.
� Traitement héparinique : limite l’hypercoagulation mais peut aggraver le syndrome hémorragique s’il existe une thrombopénie et un déficit en facteurs de coagulation.
� Indications
� C.I.V.D. sans déficit majeur en facteurs « consommable : Héparinothérapie»
� C.I.V.D. avec déficit majeur en facteur consommables Correction des déficits par transfusion de plaquettes et ou de PFC puis héparinothérapie
� Une surveillance régulière doit donc être instituée afin d’adapter le traitement
52
Hyperfibrinolyse
53
Hyperfibrinolyse pure � Primitive (rare) :
� ↑ t-PA sans fibrine � chirurgie urologique, utérine, pulmonaire, cardiaqu e, néoplasies pancréas,
prostate
� Secondaire à la formation de fibrine, modérée
� Biologie: � chute du fibrinogène < 0,8 g/L� diminution du temps de lyse des euglobulines� Taux de plaquetyes normal� PDF augmentée sans D Dimères
� Antifibrinolytiques� Aprotinine (Trasylol®)� Ac tranexamique (Exacyl®)
54
Insuffisance hépato cellulaire
� Insuffisance hépatique� Aiguë :
� hépatite fulminante � Intoxication amanite phalloïde � Toxique industriel , médicamenteux
� Chronique� Intoxication alcoolique� Hépatite C, B
� Fréquente� Trouble hémostase proportionnel à gravitéFacteur pronostic
� TP ou Facteur V <20 % : Greffe en urgence

10
55
Insuffisance hépato cellulaire� Insuffisance hépatique légère
� baisse du taux de prothrombine et des facteurs II, VII et X dont les valeurs sont comprises entre 40 et 60 %.
� Insuffisance hépatique modérée� TP, facteurs II, VII et X compris entre 30 et 50 %.� Il apparaît une baisse modérée du fibrinogène et des plaquettes.
� Insuffisance hépatique sévèreTP et facteurs II, VII et X inférieurs à 30 %. Fibrinogène est inférieur à 1g/litre. Plaquettes inférieur à 100 G/L.Il s’associe souvent des signes de C.I.V.D chronique.
� TP, facteur V < 20 %: critères de mauvais pronostic�
56
Insuffisance hépato cellulaire
� Le traitement des accidents hémorragiques ou leur prévention repose� Perfusion de PFC� Traitement hémostatique locaux.
� Attention Danger: la perfusion isolée de PPSB � Entraîne un déséquilibre entre activateur et inhibiteur de la coagulation
� risque de déclencher une CIVD
57
Anticoagulants circulantsAntiphospholipide
Antiphospholipide (antiprothrombinase, type lupique)� Très fréquent� Rencontré dans :
� Maladie de système tel que lupus érythémateux disséminé� Pathologie infectieuses de l’enfant
� Pas de signes cliniques hémorragiques sauf si thrombopénie associée
� Risque thrombotique � Biologie : TCA allongé, TP Normal
� Attention Danger: TCA allongé en pré opératoire exploration en urgence de cette situation:� Anticoaulant circulant sans risque hémorragique � Déficit en facteur VII;, IX, XI, XII 58
Anticoagulants circulantsdirigé contre un facteur
� anti facteur VIII, anti facteur V spontané� Très rare
� Rencontré dans :� Maladie de système tel que lupus érythémateux disséminé
� idiopathique
� signes hémorragiques majeurs
� Traitement � Traitement spécifique
� Immuno suppreseur
� +/- subtitution en facteur
59
Troubles hémorragiques dus àdes Médicaments interférant
sur la coagulation:
60
Troubles hémorragiques dus à des Médicaments
� Première cause de iatrogénie
� AVK� Sujet âgé , mauvaise observance, insuffisance de contrôle,
INR > 5
� Hématome sous duraux , hémorragies digestives
� Héparines � Traitement prolongé, insuffisance rénale
� Saignement post chirurgicaux, hémorragies digestives
� Anti inflammatoires� hémorragies digestives
� Chimiothérapies � Thrombopénies

11
61
Situation préopératoire
Dépistage d’une anomalie de la coagulation
62
Interrogatoire (1)
� Antécédents� Personnels
� Chirurgicaux� NON chirurgicaux
� Familiaux
� Prise de médicaments
� Maladie particulière
63
Interrogatoire (2)
Histoire +
Histoire*négative*non informative
Bilan orienté
Chirurgie
non à risque
Chirurgie
à risque
Bilan
minimum
Pas de bilan
?
64
Bilan biologique minimum ?
� Plaquettes
� TP
� TCA
� (Fibrinogène)
65
Orientations possibles après l'interrogatoire
Anomalies� Hémostase primaire
� Hémorragies précoces� Hémorragies cutanées
ou muqueuses� Coagulation
� Hématomes� Hémarthroses
� Fibrinolyse� Hémorragies tardives� Localisations
préférentielles (gynécologiques)
Caractère
� Héréditaire� Apparition précoce� Antécédents familiaux
� Acquis� Apparition tardive
(adulte)� Pathologieassociée
66
0,5-1 g/L30%30 %
10-20 %1-5 %0 %
50. 109/L
FibrinogèneFII,V,X
FXIFVIIFXIII
FXII, KHPM, PKPlaquettes
Taux minimumParamètre
D'après STV N° vol 7, 1995 p14
Taux minimum indicatif pour hémostase correcte

12
67
Déficit Incidence / Pop Gle Localisation gène Transmission------------------------------------------------------Fibrinogène 1:1 million 4 ARProthrombine 1:2 million 11 ARFacteur V 1:1 million 1 ARFacteur VII 1:500 000 13 ARFacteur VIII 1:10 000 X R lié à l’XFacteur IX 1:60 000 X R lié à l’XFacteur X 1:1 million 13 ARFacteur XI 1:1 million 4 ARFacteur XIII 1:2 million 6(ss-unité A) AR
et 1(ss-unité B)------------------------------------------------------
Fréquence des anomalies congénitales de l’Hémostase
68
Evaluation Préopératoire� Interrogatoire + Examen Clinique +/- Biologie
� Dépister trouble éventuel
� Risque hémorragique / Risque thrombotique� Acte (type, alitement, localisation…)� Patient (anomalie éventuelle de l’HS)� Traitements antithrombotiques� FR de thrombose
� Prophylaxie� Mise en place� Niveau















![Traumatisme crânien grave - reanesth.chu-bordeaux.fr´me-d... · Traumatisme crânien grave ... Microsoft PowerPoint - Trauma cranien.ppt [Mode de compatibilité] Author: rolquip](https://img.pdfslide.fr/doc/110x75/5b9a9e2409d3f2aa588baaa8/traumatisme-cranien-grave-me-d-traumatisme-cranien-grave-microsoft.jpg)













