Embed Size (px)
Citation preview

Ophtalmologie infantile
E. Bui Quoc
L’ophtalmologie infantile se caractérise par l’existence d’une période sensible du développement visuel,période au cours de laquelle toute altération de l’expérience visuelle est à risque d’amblyopie, c’est-à-direune fonction visuelle altérée par les altérations des propriétés anatomofonctionnelles des neuronesimpliquées dans la perception visuelle, essentiellement au niveau du cortex visuel. Ainsi, le rétablissementanatomique (s’il est possible) peut ne pas suffire chez l’enfant à rétablir une fonction visuelle normale, depar cette amblyopie fonctionnelle. La pathologie ophtalmologique est diverse. Isoler la pathologieophtalmologique infantile peut être un peu artificiel. Nous tentons dans ce panorama de faire le relevédes différentes situations auxquelles l’ophtalmopédiatre peut être confronté : troubles de réfractions,pathologies cornéennes, cristalliniennes, rétiniennes, pathologies infectieuses, inflammatoires,tumorales, neuro-ophtalmologie, pathologie oculomotrice, pathologies malformatives...© 2007 Elsevier Masson SAS. Tous droits réservés.
Mots clés : Vision ; Amblyopie ; Strabisme ; Cornée ; Cristallin ; Rétine
Plan
¶ Introduction 2
¶ Amblyopie et développement visuel 2Amblyopie 2Développement des fonctions visuelles chez l’humain 2
¶ Examen de l’enfant 3Dépistage 3Différentes étapes de l’examen ophtalmologique 4
¶ Troubles de la réfraction 5Définitions 5Étude de la réfraction 5
¶ Pathologie de surface 5Conjonctivites infectieuses 5Conjonctivites virales 6Conjonctivites allergiques 6Sécheresse oculaire 6Autres atteintes conjonctivales : syndrome de Lyell,atteintes dermatologiques... 6
¶ Pathologie cornéenne 7Kératites 7Dystrophies héréditaires de cornée 7
¶ Glaucome et dysgénésies du segment antérieur 7Diagnostic 8Surveillance 8Formes cliniques 8
¶ Pathologie cristallinienne et zonulaire 9Cataracte congénitale et infantile 9Traitement de la cataracte congénitale 9Microsphérophaquie 9Luxation et subluxation du cristallin 9Cataracte dans le cadre d’une persistance du vitré primitifou d’une persistance de l’artère hyaloïdienne 10
¶ Pathologie rétinienne 11Rétinopathie des prématurés 11Rétinopathies pigmentaires 11
Pathologies vasculaires 12Dysplasies rétinovitréennes 13Dyschromatopsies héréditaires 14Maladie de Stargardt ou fundus flavimaculatus 14Malformations rétiniennes et du nerf optique : « morning glorysyndrome », colobomes du nerf optique et de la rétine,fossette colobomateuse 15
¶ Strabismes 15
¶ Neuro-ophtalmologie 17Retard de maturation visuelle ou syndrome de Beauvieux 17Nystagmus 17Neuropathies optiques 17Pathologie pupillaire 18Diplopie aiguë 19
¶ Pathologie orbitopalpébrale 19Malformations orbitaires 19Malformations palpébrales et ptosis congénital 19Voies lacrymales 19Conduite à tenir devant un chalazion et devant un orgelet 20
¶ Pathologie tumorale 20Rétinoblastome 20Tumeurs orbitaires 21Hémangiomes de la face 21
¶ Traumatologie 21Conduite à tenir devant une contusion oculaire 21Fracture du plancher de l’orbite 22Plaies transfixiantes du globe 22Plaie de paupière 22Brûlures oculaires 22
¶ Uvéites 23Uvéites infectieuses : herpès, cytomégalovirus, toxoplasmose,toxocarose... 23Arthrite juvénile idiopathique 23Autres étiologies d’uvéite 23
¶ Conclusion 23
¶ 4-120-A-10
1Pédiatrie
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
181920
212223
242526
27282930313233
343536
37383940
41424344454647
484950
51525354555657
58
596061626364
6566676869
70717273
747576777879
8081828384
8586

■ IntroductionTraiter (au sens de disserter) d’ophtalmologie infantile en
quelques mots, ou quelques pages, est une tâche difficile pourde multiples raisons. L’ophtalmologie en soi est une disciplined’une diversité étonnante ; certes, la pathologie réfractive est leplus commun motif de consultation, mais il est de nombreusessituations où le défaut visuel requiert bien plus qu’une simplecorrection optique. La sur-spécialisation devient la règle,souvent sur un support anatomique. On retrouve des ophtal-mologistes spécialistes de la cornée (avec les variantes de lacontactologie, de la chirurgie réfractive ou encore des patholo-gies de la surface oculaire). Il existe des chirurgiens experts enpathologie du cristallin et à la pointe des dernières innovationsen chirurgie de la cataracte. D’autres sont des passionnés depathologie rétinienne, avec souvent la distinction entre « réti-nologues » médicaux (traitant les pathologies vasculaires, ladégénérescence maculaire liée à l’âge, la rétinopathie diabétique)et « rétinologues » chirurgicaux (dont l’activité englobe lachirurgie du décollement de rétine, des pathologies maculairescomme les membranes épirétiniennes ou les trous maculaires...).Certains étendent leur pré carré en dehors de l’œil et s’exercentà la pathologie orbitopalpébrale. D’autres classifications de sur-spécialités existent : les neuro-ophtalmologues s’éloignent del’œil pour se rapprocher du cortex, en longeant les voiesvisuelles ; les « uvéitologues » traitent de pathologie inflamma-toire et infectieuse oculaire, avec le soutien précieux de leurscollègues internistes ; les strabologues traitent les désordresoculomoteurs ; les « glaucomatologues » exercent leur art entraitant (médicalement et chirurgicalement) ces divers facteurs(hypertonie oculaire, altérations vasculaires...) qui peuvent êtreresponsables de la neuropathie optique cécitante, autrementappelée « glaucome »...
Quelle est – au sein de ces différents groupes – la place del’ophtalmologie dite pédiatrique ou infantile. L’ophtalmopédia-tre focalise son exercice en fonction de l’âge du patient, et doitgérer les différents domaines suscités correspondants à la quasi-totalité de la pathologie ophtalmologique. La sur-spécialité« ophtalmologie infantile » est-elle donc pertinente ? Oui, biensûr, mais l’ophtalmopédiatre doit avoir une vue globale de ladiscipline, doit pouvoir traiter adultes et enfants, tant il existeun continuum pathologique tout au long de la vie : citonsuniquement l’exemple du gène ABCR, dont les différentsgénotypes peuvent être responsables d’un phénotype « maladiede Stargardt » chez un enfant mais aussi de certaines formes dedégénérescence maculaire liée à l’âge chez la personne plusâgée [1]. Tant et si bien que l’examen des parents est – dansquelque domaine de l’ophtalmologie infantile que ce soit – toutà fait fondamental !
Comment maintenant exposer avec une certaine clartédidactique cette vaste pathologie infantile ? Après un rappelphysiologique sur l’amblyopie et la période sensible du déve-loppement visuel, la classification adoptée dans cette disserta-tion, qui se veut à la fois exhaustive mais nécessairementrésumée et condensée, reprendra en partie le schéma anatomi-que correspondant à une explication physiologique très simple :pour voir correctement, il faut que les rayons lumineux arriventprécisément sur la rétine (avec une correction optique d’amé-tropie le cas échéant), et aient traversé des milieux clairs etparfaitement transparents : cornée, chambre antérieure, cristal-lin, vitré ; puis il faut que la rétine fonctionne, que le nerfoptique fonctionne, et que l’intégration perceptive corticale soitassurée correctement (Fig. 1). Cela ramène l’ophtalmologie àune spécialité très simple : l’ophtalmologiste recherche, àchaque niveau de transmission de l’information sensorielle(l’image), une anomalie pouvant expliquer la mauvaise vision.Outre cette classification seront traités bien évidemment lesnombreux autres aspects de la pathologie ophtalmologiqueinfantile, en restant conscients qu’il serait fait silence, au coursde cet exposé forcément bref, de certaines pathologies parmi lesplus rares.
■ Amblyopie et développementvisuelAmblyopie
Chez le mammifère supérieur, le développement anatomo-fonctionnel des structures oculaires, des voies visuelles et deszones cérébrales impliquées dans la perception visuelle n’est pasachevé à la naissance. C’est pourquoi une altération précoce del’expérience visuelle chez l’enfant, quelle que soit la pathologieoculaire en cause, est à risque d’amblyopie. Cette pathologiepeut se définir en première approximation, à l’aide du diction-naire Bailly [2], comme une vue (η ωp η) faible (αµbkE ιa). Ledictionnaire Garnier-Delamare [3] évoque quant à lui une« diminution de l’acuité visuelle ».
L’amblyopie, fonctionnelle ou organique (cette distinctiondemeure utile dans un but didactique bien que les frontièressoient labiles entre ces deux formes d’amblyopie), est unepathologie qu’il est indispensable de traiter le plus précocementet le plus énergiquement possible.
Le risque clinique de développement d’une amblyopie estconnu depuis longtemps, en particulier en cas de strabisme [4].Toute pathologie oculaire pédiatrique doit donc avoir à la foisl’objectif de diagnostiquer cette pathologie, traiter cette patho-logie et rétablir les structures oculaires dans des conditionsoptimales, et l’objectif de « rééduquer » l’amblyopie le caséchéant.
Les techniques de « rééducation » de l’amblyopie et lesprotocoles utilisés sont multiples (citons pour mémoire letraitement de référence que constitue l’occlusion totale, quecomplètent les pénalisations optiques ou pharmacologiques),efficaces si bien utilisés [5] bien que ces moyens peuvent paraîtrerelativement simples par rapport aux processus neurophysiolo-giques sous-jacents extrêmement précis.
La période sensible du développement visuel serait terminéeentre 6 et 8 ans, marquant le terme de cette période « critique »au cours de laquelle il existe un risque majeur d’amblyopiefonctionnelle [6].
Développement des fonctions visuelleschez l’humain
Comme chez les autres mammifères, les fonctions visuellesdu nouveau-né humain ne sont pas d’emblée optimales. Ce
1 2 3 4
5
8910 7
6
Figure 1. Anatomie simplifiée de l’œil. 1. Cornée ; 2. sclère ; 3. cho-roïde ; 4. rétine ; 5. nerf optique ; 6. vaisseaux sanguins ; 7. macula ; 8.cristallin ; 9. pupille ; 10. iris.
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103104105106107108109110111112113114115116117118
119120121122123124125126127128129130131132133134
135136137138139140141142143144145146147148149150151152153154155156
157
158
159
160161162163164165166167168169170171172173174175176177178179180181182183184185186187188189190191192
193
194
195196
4-120-A-10 ¶ Ophtalmologie infantile
2 Pédiatrie

n’est qu’avec l’âge que celles-ci se développent. De nombreusesétudes d’estimation de ces fonctions visuelles à différents âges,par des tests d’acuité visuelle monoculaire, des tests de visionbinoculaire mais aussi d’autres tests psychovisuels peuventpermettre d’évaluer le profil chronologique de développementde la fonction visuelle chez l’humain. Il faut souligner ladifficulté de l’étude de la fonction visuelle, du fait de lamultiplicité même de l’éventail des paramètres étudiés : acuitévisuelle, vision des couleurs, champ visuel, fonction de sensibi-lité aux contrastes...
Développement de l’acuité visuelle
L’évolution de l’acuité visuelle normale est difficile à évaluerchez l’enfant. Sa mesure est estimée, le plus souvent par destests cliniques (bébé-vision à l’aide de cartons de Teller avantl’âge de 18 mois), mesure de l’acuité visuelle par lectured’optotypes (échelle de Pigassou), à partir de 3 à 4 ans. Lesdifférents auteurs s’accordent pour estimer que l’acuité visuelleest de 1/10 à 3 mois, 3/10 à 1 an, 10/10 à 4 ans [7].
Développement de la vision binoculaire
La mesure de la vision binoculaire dépend du test utilisé :tests de stéréoscopie qualitative reposant sur la notion deparallaxe stéréoscopique (vues d’un même objet selon un angledifférent par les deux yeux, créant ainsi une disparité réti-nienne) ; tests de stéréoscopie fine, constitués de deux stéréo-grammes superposés avec des nappes de points aléatoires.
Chez l’adulte, la stéréoscopie normale est inférieure à50 secondes d’arc.
Chez l’enfant, l’évaluation peut être faite selon la méthodedu regard préférentiel, avec des stéréogrammes. On considèreque la stéréoscopie est absente dans les premiers mois de viepour apparaître, brutalement, entre le 3e et le 5e mois [8]. Ellen’atteint cependant des valeurs stables et de profil « adulte »qu’après 6 ans, comme l’a montré Romano. Ses travaux ontporté sur l’étude, par le stéréotest de Titmus, de la stéréoscopiede 321 enfants de 1,5 à 13 ans, considérés comme ayant desfonctions visuelles normales, c’est-à-dire sans anomalie ophtal-mologique ou antécédents à risque d’altérer ou d’avoir altéré lastéréoscopie (cet « a priori » est un facteur limitant la validité decette étude par ailleurs intéressante). La stéréoscopie atteint à5 ans 140 secondes, à 6 ans 80 secondes, à 9 ans 40 secondes [9].
Autres moyens d’étude du développementde la fonction visuelle
L’examen du champ visuel, qu’il soit statique ou dynamique,est bien entendu impossible chez le petit enfant, en pratiqueréalisable à partir de 6 ou 7 ans. On peut cependant l’estimeren étudiant les saccades consécutives à des stimuli périphéri-ques, en nasal ou en temporal. Ainsi, il a été estimé à 30° ennasal comme en temporal à la naissance, pour n’atteindre unetaille presque adulte qu’à la fin de la première année [10].
La fonction de sensibilité au contraste de même ne peutqu’être estimée chez l’enfant à partir de déductions sur ledéveloppement des canaux de codage de fréquence spatiale(couples On/Off de cellules ganglionnaires). Ce n’est qu’à
3 mois que la fonction de sensibilité au contraste atteindrait unprofil adulte (profil d’une courbe « en cloche »), avec cepen-dant :• un décalage vers les basses fréquences spatiales, corrélative-
ment à l’acuité visuelle qui demeure basse à cet âge ;• un décalage vers le bas de la sensibilité au contraste, qui
demeure plus faible jusqu’au début de l’adolescence [11, 12].Les potentiels évoqués visuels (PEV) chez l’enfant doivent être
interprétés avec précaution. Les résultats dépendent bienévidemment de la maturation rétinienne, de la myélinisationdes voies optiques. Ils doivent être systématiquement comparésau bruit de fond électrique. Pour les potentiels évoqués station-naires, l’amplitude maximale recueillie va dépendre de lafréquence temporelle de stimulation : 2 Hz à la naissance, 4 Hzà 6 mois, 6 Hz à 10 mois, 10 Hz après 12 mois. Les potentielsévoqués par damiers permettent une évaluation de la discrimi-nation spatiale mais nécessitent une fixation. Ainsi, l’interpré-tation des PEV dans le développement de la fonction visuelleest difficile. Rappelons que du fait de la magnification macu-laire, les PEV reflètent la fonction maculaire et la conductiondes voies visuelles. Teller rappelle que, pour la majorité desauteurs, les PEV montreraient un profil adulte à 6-8 mois [13].
■ Examen de l’enfant
DépistageLe dépistage des affections ophtalmologiques est une néces-
sité du fait de l’existence d’une période sensible du développe-ment visuel : le pronostic fonctionnel dépend, en casd’anomalie asymétrique entre les deux yeux, de la précocité dela thérapeutique mise en œuvre car la plasticité cérébrale, quece soit dans le sens d’un développement pathologique, maisaussi dans le sens d’une possibilité de récupération fonction-nelle, est limitée après l’âge de 6 ans. La question se pose doncdes moyens de dépistage des anomalies ophtalmologiquesinfantiles [14-16]. Le nouveau carnet de santé insiste sur ledépistage des troubles sensoriels chez l’enfant, en particulier lespathologies oculaires. L’étude des sérologies rubéole et toxo-plasmose recherche une contamination maternofœtale, ces deuxgermes pouvant causer des rétinopathies néonatales. L’étudedans les tout premiers mois de vie d’une anomalie de la cornée(taille, transparence), recherche à dépister un glaucome congé-nital. La modification de la lueur pupillaire peut être causée parune cataracte congénitale, mais aussi le redoutable rétinoblas-tome. Un strabisme comme un nystagmus peuvent être dessymptômes de malvoyance, et leur survenue nécessite desexplorations spécialisées. Ce n’est qu’à partir de l’examen de latroisième année qu’on commence à évoquer l’acuité visuelle, lavision des couleurs (fondamentale chez le garçon, en particulierpour l’orientation professionnelle), la vision stéréoscopique. Ilest à noter que nulle part n’est évoquée la réfraction. Sa mesureest bien entendu liée à l’acuité visuelle, mais il est certain quele dépistage d’amétropies précoces, en particulier des myopiesfortes ou des anisométropies (différence de réfraction entre lesdeux yeux), serait souhaitable. Il est possible dès la premièreannée de vie et grandement facilité par l’autoréfractomètreportable, peut-être plus difficile entre 1 et 3 ans, car l’enfant estplus pusillanime, mais tout à fait facile après. Le problème est,nous l’avons évoqué, la nécessité d’une cycloplégie, donc celaprend du temps, et la nécessité de faire l’examen par unophtalmologiste. Les éléments du carnet de santé ont été établispour qu’il soit réalisable par le pédiatre ou le médecin généra-liste. Son rôle est fondamental, et au moindre doute, un avisspécialisé doit être requis. Enfin il faut le rappeler, le bébé-vision ne sert à rien en dépistage et ne sert qu’à faussementrassurer le médecin et les parents !
En France, les examens, inscrits sur le carnet de santé, sontréalisés par le médecin généraliste ou le pédiatre. On peutrecommander un examen ophtalmologique avant l’âge de 1 an,vers l’âge de 3 ou 4 ans (l’enfant peut lire les optotypes), etavant l’entrée en cours préparatoire, où un examen completpeut être réalisé. Il comprend : acuité visuelle de loin et de prèssans et avec correction, avec la formule réfractive, un examen
“ Point fort
Diagnostic de l’amblyopie « monoculaire ».• Différence d’acuité visuelle entre les deux yeux > deuxlignes.• Amblyopie profonde, acuité visuelle ≤ 1/10e.• Amblyopie moyenne, acuité visuelle entre 1 et 4/10e.• Amblyopie légère, acuité visuelle > 4/10e.• L’acuité visuelle est le paramètre utilisé en pratique pourdiagnostiquer et suivre une rééducation d’amblyopie.
197198199200201202203204205206
207
208209210211212213214
215
216217218219220221222223224225226227228229230231232233234235236
237
238
239240241242243244245
246247248249
250251252253254255256257258259260261262263264265266267268269270271
272
273
274275276277278279280281282283284285286287288289290291292293294295296297298299300301302303304305306307308309310311312313314315316317318319320
.
Ophtalmologie infantile ¶ 4-120-A-10
3Pédiatrie

du fond d’œil, un examen de la vision binoculaire, la recherched’un strabisme, d’un nystagmus, d’une poursuite oculairenormale, d’autres anomalies ophtalmologiques.
Il est certes difficile d’appliquer cette règle stricte et depratiquer systématiquement ces examens successifs. La vigilanceest particulièrement nécessaire en cas d’anomalie patente oud’antécédents personnels ou familiaux significatifs. L’examenpostnatal par le pédiatre est fondamental, afin de dépister desanomalies congénitales, en particulier le glaucome congénitalqui est une véritable urgence ; nous y reviendrons.
Différentes étapes de l’examenophtalmologique
InterrogatoireL’âge d’apparition d’une éventuelle anomalie ayant amené à
consulter (strabisme, leucocorie, larmoiement, mauvais com-portement visuel, baisse de vision...) est fondamental. L’interro-gatoire des parents est indispensable. L’interrogatoire permetégalement de préciser les antécédents de l’enfant, à la recherche
d’une pathologie au cours de la grossesse, d’une prématurité quiest reconnue comme un facteur de risque de strabisme. Lesantécédents familiaux sont notés.
InspectionL’inspection recherche une dysmorphie et une pathologie
associée. Il faut se rappeler qu’un épicanthus, c’est-à-dire unrepli palpébral interne, peut mimer un strabisme, en particulierdans le regard latéral ; il peut cependant coexister avec uneréelle déviation des yeux.
Bilan sensoriel
Appréciation de l’acuité visuelle
Le comportement visuel de l’enfant est observé. Chez lenourrisson est apprécié l’intérêt visuel, le sourire aux visagesfamiliers, le suivi des objets et la captation du regard. La mesurede l’acuité visuelle par lecture d’optotypes, possible et fiable engénéral à partir de 3 à 4 ans, doit être réalisée de loin et de près,avec et sans correction La correction optique est établie aprèsexamen sous cycloplégie (nous y reviendrons).
Les tests de « bébé-vision », à l’aide de carton de Teller, baséssur le principe du regard préférentiel, sont possibles jusqu’à18 mois environ. Le nourrisson oriente son regard préférentiel-lement vers un réseau structuré s’il le distingue au sein d’uneplage uniforme. Le « bébé-vision » n’est en aucun cas un bontest de dépistage de l’amblyopie [17]. Il est exceptionnellementutile en cas de strabisme, sauf en cas de suspicion d’amblyopieassociée à un microstrabisme. Il peut aussi être indiqué pour lesuivi d’une amblyopie traitée.
Un test très simple mais très utile à la recherche d’uneamblyopie chez le nourrisson est le test d’occlusion d’un œil,avec le doigt ou avec un cache : si l’œil sain est couvert, l’enfantamblyope va chercher à enlever le cache, ce qui est moinsprobable si c’est l’œil amblyope qui a été caché ou bien toutsimplement en l’absence d’amblyopie.
La mesure de l’acuité visuelle reste délicate entre 18 et36 mois. On estime que l’enfant voit 1/10e à 1 mois, 2 à 3/10e
à 1 an, 5/10e à 4 ans et 10/10e à 6 ans. L’isoacuité, c’est-à-direune acuité visuelle égale après correction, entre les deux yeux,doit être la règle quel que soit l’âge.
Étude de la vision binoculaire
Après l’évaluation de la vision monoculaire, et seulement sicelle-ci est satisfaisante et égale entre les deux yeux, il fautévaluer la vision binoculaire au moyen de deux types de tests :les tests de stéréoscopie qualitative (tests de Wirt et Titmus, testde Randot) et les tests de stéréoscopie quantitative constitués dedeux stéréogrammes superposés avec des nappes de pointsaléatoires, comme le TNO. Le test de Lang de type I, égalementà base de points aléatoires, peut être possible dès 2 ans, l’enfantmontrant l’image « en relief » s’il la distingue.
Bilan moteurL’examen des reflets cornéens est le seul examen moteur
praticable chez le nourrisson : le reflet cornéen d’une lumièreréfléchie, par exemple un ophtalmoscope, doit être centré. Encas de strabisme divergent ou exodéviation, le reflet est décaléen nasal. En cas de strabisme convergent ou ésodéviation, lereflet est décalé en temporal. En cas de strabisme vertical, lereflet est décalé en supérieur ou en inférieur.
Les tests de l’écran sont possibles dès 2 ou 3 ans (dès quel’enfant fixe bien). Il s’agit des tests de référence : test de l’écranunilatéral et test de l’écran alterné. La déviation est étudiée envision de loin (fixation d’un point lumineux ou d’une cible à5 m) et en vision de près (fixation d’une cible à 33 cm). Il fautcommencer par le test de l’écran unilatéral pour ne pas démas-quer une hétérophorie par le test de l’écran alterné qui estdissociant et rompt la fusion. En interposant des prismes devaleurs croissantes devant un œil, on peut apprécier et quanti-fier la déviation oculaire, mesurée en dioptries prismatiques (àne pas confondre avec les dioptries optiques des amétropies).
De nombreux autres moyens d’études sont utilisés. Citons lesynoptophore qui permet d’étudier l’angle objectif et l’angle
“ Point fort
Examen ophtalmologique de l’enfant, adapté àl’âge et la coopération.• Interrogatoire, généalogie, antécédents (grossesse,prématurité).• Inspection : dysmorphie, comportement visuel,nystagmus.• Bilan moteur :
C reflets cornéens ;C test de l’écran unilatéral puis test de l’écran bilatéral
ou alterné, (synoptophore) ;C motricité oculaire conjuguée (fixation, saccades,
poursuite) ;C motilité oculaire (ductions, versions, vergences).
• Bilan sensoriel :C réfraction objective, skiascopie sous cycloplégie
(savoir répéter la réfraction sous cycloplégieplusieurs fois par an le cas échéant) ;
C test d’occlusion unioculaire, acuité visuellemonoculaire, acuité visuelle brute, test 4D basetemporale de Jampolsky ou biprisme de Gracis ;
C pas d’intérêt du « bébé-vision » dans le dépistage del’amblyopie (cf. Bourcier-Bareil et al. 2001).
• Examen ophtalmologique des structures oculaires :segment antérieur, TO, segment postérieur.
“ Point fort
Examens de dépistage : ce qui peut être raison-nablement préconisé.• Examen pédiatrique en postnatal immédiat :fondamental !• Examen (ophtalmologique) de l’enfant avant 1 an.• Examen ophtalmologique vers 3 ans (lorsque l’enfantfixe bien et peut lire les optotypes de Pigassou ou deCadet), avant l’entrée en cours préparatoire au moins,avec : bilan anatomique avec fond d’œil, bilan sensoriel,bilan moteur.
321322323
324325326327328329330
331
332
333
334335336337338
339340341
342
343344345346347
348
349
350351352353354355356357358359360361362363364365366367368369370371372373374375376
377
378379380381382383384385386
387
388389390391392393394395396397398399400401402403404405406407
.
4-120-A-10 ¶ Ophtalmologie infantile
4 Pédiatrie

subjectif, d’apprécier la correspondance rétinienne normale ouanormale, c’est-à-dire la correspondance entre le point defixation et le centre de la fovéola (région centromaculaire).
La motricité oculaire est étudiée dans les neuf positions duregard : étude des mouvements de duction (chaque œil estétudié séparément) et des mouvements de version (les deuxyeux sont étudiées de façon simultanée) à la recherche d’hype-ractions ou d’hypoactions musculaires des six muscles oculo-moteurs. Les mouvements conjugués des yeux ou vergencessont aussi étudiés. L’étude de l’oculomotricité est parfoisprécisée par l’examen coordimétrique, à l’aide de l’appareil deHess-Lancaster. Il faut rechercher un nystagmus, un torticolisassociés.
Examen ophtalmologique des structures oculaires
L’examen ophtalmologique précise l’intégrité des structuresde l’œil : conjonctive, cornée, chambre antérieure, iris. Aprèsdilatation pupillaire, la transparence cristallinienne est appré-ciée, la cavité vitréenne étudiée, la rétine examinée avec uneattention particulière à l’examen du nerf optique et de la régioncentrale maculaire.
Chez le grand enfant à partir de 3 à 4 ans, cet examenanatomique peut être réalisé au biomicroscope (lampe à fente).L’enfant est assis sur les genoux de ses parents ou bien est lui-même à genoux sur le fauteuil d’examen. Chez le plus petit,l’enfant est allongé, maintenu (ce qui est particulièrementdifficile entre 18 et 36 mois), et l’examen est réalisé à l’ophtal-moscope direct, après éventuelle mise en place d’un écarteur àpaupière, sous anesthésie locale topique.
Le tonus oculaire, mesuré avant dilatation, est appréciableaprès 3 ou 4 ans, au tonomètre de Goldmann ou au tonomètreà air. Avant, l’anesthésie générale est nécessaire, avec mesure autonomètre portable de Perkins, en tenant compte de l’anesthésiequi diminue de 30 à 50 % les valeurs tensionnelles.
L’examen sous anesthésie générale est parfois nécessaire, encas de doute diagnostique, ou encore juste avant une chirurgieréalisée dans le même temps anesthésique.
Examens complémentaires
L’examen du champ visuel (statique automatisé ou cinétiqueà la coupole de Goldmann) n’est uniquement possible qu’àpartir de 6 ans environ. Le test psychophysique de la fonctionde sensibilité aux contrastes est possible dès 5 ou 6 ans. Le bilanélectrophysiologique comprend l’électro-oculogramme sensoriel(EOGs) qui teste la fonction de l’épithélium pigmentairerétinien (il nécessite une coopération et n’est pas réalisableavant 4 à 5 ans), l’électrorétinogramme qui teste la fonction dela neurorétine, et les PEV, qui testent de façon globale à la foisla fonction maculaire et la fonction de conduction le long desvoies optiques jusqu’au cortex visuel. Si l’électrorétinogrammeréalisé sous anesthésie est souvent fiable, les PEV sous anesthésiesont ininterprétables, car l’anesthésie en elle-même modifie trèsfortement la réponse évoquée. Le test de vision des couleurs estun examen très simple : le test d’Ishiara dépiste les dyschroma-topsies héréditaires, le test du 15-Hue saturé et désaturé teste lesdyschromatopsies acquises.
■ Troubles de la réfraction [18]
DéfinitionsUne amétropie est un trouble réfractif : le dioptre oculaire –
approximativement défini par la combinaison du dioptrecornéen d’une puissance des deux tiers de la puissance totale,soit 44 dioptries en moyenne, et du dioptre cristallinien d’unepuissance du tiers de la puissance totale, soit 22 dioptries enmoyenne – ne permet pas la focalisation des rayons lumineuxincidents sur la rétine, mais en avant ou en arrière. L’imageperçue n’est pas nette. On distingue les amétropies axiles(longueur de l’œil inadaptée à la puissance du dioptre oculaire),les amétropies d’indice (l’indice de réfraction des milieux – engénéral le cristallin – varie, ce qui modifie la puissance normale
du dioptre), les amétropies de courbure, par modification de lacourbure de la cornée, modifiant de ce fait la puissance dudioptre cornéen.
En cas de myopie, l’image d’un objet situé à l’infini (assimi-lable à une distance de 5 m en pratique) se projette en avantde la rétine. Cette amétropie se corrige par un verre concave(divergent).
En cas d’hypermétropie (hyperopie), l’image d’un objet situéà l’infini se projette en arrière de la rétine. Cette amétropie secorrige par un verre convexe (convergent). Le pouvoir d’accom-modation du cristallin, c’est-à-dire sa capacité à augmenter sapuissance par modification de sa courbure sous l’effet de lacontraction des muscles ciliaires, est fort chez l’enfant etdiminue avec l’âge ; il permet de compenser une hypermétropieen ramenant le rayon lumineux vers l’avant. Cette compensa-tion peut se manifester par des troubles fonctionnels à type decéphalées intermittentes par exemple, ou par un strabismeconvergent, du fait du réflexe d’accommodation-convergence.
L’anisométropie est une variation importante de l’amétropiedes deux yeux. Elle peut induire une amblyopie de l’œil le plusamétrope.
L’astigmatisme correspond à une différence de puissance dudioptre oculaire selon deux méridiens différents. Elle est le plussouvent cornéenne : la courbure de la cornée est plus impor-tante selon son méridien vertical que selon son méridienhorizontal dans un astigmatisme dit conforme. L’image d’uncercle correspond à une ellipse.
Étude de la réfractionLa mesure objective de la réfraction est possible même chez
le tout-petit par la méthode de la skiascopie, c’est-à-dire l’étudedu mouvement d’une barre lumineuse reflétée dans l’airepupillaire. L’examen se pratique obligatoirement après relâche-ment de l’accommodation par instillation d’un collyre cycloplé-gique. L’atropine est le « gold standard » ; elle s’utilise à la dosed’une goutte matin et soir pendant la semaine précédantl’examen.
Le dosage varie en fonction de l’âge : atropine à 0,3 % avant3 ans, à 0,5 % entre 3 et 5 ans, à 1 % au-delà de 5 ans. Lesalternatives sont l’homatropine 1 %, instillée toutes les20 minutes 2 heures avant l’examen, ou le cyclopentolate(Skiacol®), dont les contre-indications sont les antécédentsneurologiques en particulier d’épilepsie non traitée et stabilisée,le syndrome de Down, l’âge inférieur à 1 an. Ce collyre est parailleurs peu efficace chez le mélanoderme. Le cyclopentolate estinstillé trois fois en un quart heure et l’examen est réalisé à la45e minute (effet éphémère de 1 h). L’examen sous cycloplégiepermet de mettre au repos les phénomènes excessifs d’accom-modation. Le principe est l’observation du mouvement de lafente lumineuse, en sens direct ou inverse, et la recherche de lapuissance du verre permettant d’inverser ce mouvement,correspondant à l’amétropie (en tenant compte de la distanced’examen). La différence de mesure entre deux méridiensperpendiculaires permet la mesure de l’astigmatisme. La mesurede la réfraction a été grandement facilitée par les réfracteursautomatiques, et en particulier les réfracteurs portatifs chez lenourrisson. Il faut savoir renouveler l’étude de la réfraction etles cycloplégies, car l’état réfractif n’est pas une donnée figée ;elle est au contraire évolutive. La correction optique nécessaireà l’enfant peut varier au cours du temps.
■ Pathologie de surface
Conjonctivites infectieuses [19]
La pathologie conjonctivale infectieuse est fréquente chezl’enfant, correspondant à un œil « sale » avec hyperhémieconjonctivale et présence de sécrétions le matin. Les germes leplus souvent en cause sont le streptocoque, le staphylocoque, lepneumocoque. Le traitement associe un lavage oculaire matin etsoir et une antibiothérapie locale à « forte dose », c’est-à-dire aumoins 5 gouttes par jour pendant une courte période de 5 à
408409410411412413414415416417418419420
421
422423424425426427428429430431432433434435436437438439440441442443
444
445446447448449450451452453454455456457458459460461
462
463
464465466467468469470471472473474
475476477478479480481482483484485486487488489490491492493494495496497498499500501
502
503504505506507508509510511512513514515516517518519520521522523524525526527528529530531532533
534
535
536537538539540541542
Ophtalmologie infantile ¶ 4-120-A-10
5Pédiatrie

6 jours. On peut utiliser par exemple la rifampicine (Rifamy-cine®) ou la tobramycine (Tobrex®).
Les récidives infectieuses au cours des premiers mois de viedoivent faire évoquer une imperforation des voies lacrymales.On peut se contenter jusqu’au sixième mois de traiter lesépisodes infectieux de façon itérative, puis, en cas d’échec, deproposer un sondage des voies lacrymales, qui peut se réaliseravant le douzième mois en consultation, avec une simplecontention. Au-delà, l’anesthésie générale est requise, avec posed’une sonde en silicone dans les voies lacrymales de façontransitoire, qui est ôtée après 2 ou 3 mois. L’indication d’unedacryocystorhinostomie est rare ; par exemple après un épisodede dacryocystite.
La conjonctivite néonatale gonococcique, rare et sévère,caractérisée par un chémosis important et un œdème palpébralsurvenant dans les premiers jours de vie, est prévenue parl’instillation chez le nouveau-né d’un collyre au nitrate d’argentà 1 %.
Rappelons que Chlamydia trachomatis peut être responsabled’une conjonctivite néonatale de début plus tardif (5e au 10e
jour), d’aspect aussi inflammatoire que la conjonctivite gono-coccique. Plus tard, ce germe peut être responsable d’uneconjonctivite folliculaire, d’évolution volontiers chronique, etpouvant laisser des séquelles à type de néovascularisationlimbique.
Conjonctivites virales [20]
Les conjonctivites virales prennent l’aspect d’une conjoncti-vite folliculaire, avec peu de sécrétions, et parfois uniquementlimitées à une hyperhémie conjonctivale uni- ou bilatérale.Nous citerons uniquement les conjonctivites à adénovirus et lesconjonctivites herpétiques.
La conjonctivite à adénovirus survient le plus souvent dansun contexte épidémique, avec notion de contage. Il s’agit d’uneconjonctivite folliculaire dans laquelle la relative pauvreté dessignes d’examen contraste avec une symptomatologie bruyante.Il n’existe pas de traitement spécifique ayant fait preuve de sonefficacité. Il faut proposer un simple lavage oculaire et descollyres lubrifiants.
Herpes simplex virus de type I est responsable de conjoncti-vite folliculaire banale, mais peut se compliquer de kératiteponctuée superficielle, et dans certains cas d’une réelle kératiteavec aspect de dendrite. Le traitement par aciclovir en pom-made doit, dans tous les cas, être évité du fait de la toxicitélocale de cette pommade. En cas d’atteinte cornéenne, outre letraitement lubrifiant local, on peut proposer un grattage localdes bords de l’ulcère (où se situe la réplication virale) et untraitement par aciclovir ou valaciclovir systémique pendant7 jours. La complication majeure de la kératoconjonctiviteherpétique est la récidive et la chronicisation, avec risqued’opacité cornéenne cicatricielle.
Conjonctivites allergiques [21]
Conjonctivite allergique saisonnière
C’est la plus fréquente des conjonctivites allergiques. Elletouche l’adulte et l’enfant. Il s’agit de la composante oculaire du
banal « rhume des foins ». Elle est médiée par une réponseimmunoglobulines (Ig) E à un allergène le plus souvent aéro-porté, à type de pollen, de moisissure. Les allergènes retrouvéssont certains arbres, des graminées, des plantes herbacées, desmoisissures comme Alternaria alternata. Dans sa forme aiguë, laconjonctivite allergique saisonnière entraîne des signes cliniquesbruyants avec atteinte bilatérale, prurit intense, chémosis,larmoiement. Dans sa forme chronique, il existe un prurit, unpicotement oculaire permanent, un larmoiement, une discrètekératite ponctuée superficielle, une conjonctivite papillaire.
Conjonctivite allergique perannuelleElle est beaucoup plus rare. Elle atteint également l’adulte et
l’enfant. Le symptômes sont présents toute l’année avec desrecrudescences saisonnières, d’où la confusion fréquente avec lesconjonctivites allergiques saisonnières, d’autant que les signescliniques sont identiques bien que plus discrets. Il existe unehypertrophie papillaire discrète, des follicules. Les allergènes encause sont les acariens, les blattes, les moisissures, les phanèresd’animaux. On peut retrouver des allergènes professionnels.
Kératoconjonctivite vernaleC’est un cas particulier de conjonctivite allergique, survenant
plus volontiers chez le garçon de 2 à10 ans. Il existe unlarmoiement important, une photophobie majeure. Il s’agitd’une conjonctivite sévère. L’examen retrouve un infiltratlimbique (grains de Trantas) (Fig. 2A) ; des pavés sont visiblessur la conjonctive palpébrale, pour peu que l’on s’astreigne àretourner les paupières (Fig. 2B). La plaque vernale correspondà une complication à type d’opacité cornéenne.
Traitement des conjonctivites allergiquesLe traitement des conjonctivites allergiques repose sur un
traitement de la phase aiguë avec lavage oculaire abondant ausérum physiologique ou équivalent (sans conservateur, c’est-à-dire en unidoses) 2 à 3 fois par jour, et un collyre antihistami-nique 2 à 3 fois par jour (posologie variable selon les produits),pendant 10 à 15 jours.
Le traitement au long cours repose également sur le lavageoculaire quotidien, l’éviction de l’allergène si possible, et danscertains cas, un traitement antihistaminique local au long coursest nécessaire. On peut également utiliser un antidégranulantmastocytaire (cromoglycate), qui agit en prévention aux phasesprécoces de la réaction allergique.
La kératoconjonctivite vernale pose des problèmes thérapeu-tiques. On peut être amené dans certains cas à proposer unecorticothérapie courte locale.
Sécheresse oculaireLa sécheresse oculaire vraie est rare chez l’enfant, mais elle
peut accompagner une pathologie allergique chronique. Sontraitement repose sur les collyres lubrifiants, dont il existe unelarge gamme, avec des produits de viscosité variable.
Autres atteintes conjonctivales : syndromede Lyell, atteintes dermatologiques...
L’épidermolyse nécrosante subaiguë ou syndrome de Lyellassocie une dermatose bulleuse gravissime, avec également
Figure 2.A. Kératoconjonctivite vernale. Infiltrat limbique.B. Kératoconjonctivite vernale. Pavés conjoncti-vaux.
543544545546547548549550551552553554555556557558559560561562563564565566567
568
569570571572573574575576577578579580581582583584585586587588589590591592
593
594
595596
597598599600601602603604605606
607
608609610611612613614615
616
617618619620621622623624
625
626627628629630631632633634635636637638639640
641
642643644645
646
647
648649
4-120-A-10 ¶ Ophtalmologie infantile
6 Pédiatrie

atteinte muqueuse ; elle est secondaire à une réaction à certainsagents infectieux (comme le mycoplasme) ou certains médica-ments. Au plan ophtalmologique, il existe une conjonctiviteinflammatoire chronique sévère, avec nécrose des cellules àmucines et atteinte limbique. C’est l’atteinte limbique qui grèvele pronostic, car elle empêche la cicatrisation cornéenne et peutrésulter en un amincissement cornéen, voire une perforation. Letraitement repose sur les soins locaux attentifs et réguliers :pommade ophtalmique, antibioprophylaxie locale, soins desculs-de-sac pour prévenir les symblépharons. Il peut êtreintroduit une corticothérapie locale à faible dose ou de laciclosporine en local. Au stade de complication cornéennesévère, on peut proposer une greffe de membrane amniotiquepour favoriser la cicatrisation. À distance, les éventuellesséquelles cornéennes opaques peuvent justifier d’une greffe decornée.
De nombreuses génodermatoses, comme les ichtyoses, peu-vent entraîner des complications oculaires, par atteinte palpé-brale, entraînant une malocclusion palpébrale, uneconjonctivite chronique avec souvent une kératite d’expositionpouvant résulter en des opacifications cornéennes. Le traitementlubrifiant local au long cours, certes nécessaire, est parfoisinsuffisant, et il ne faut pas hésiter à proposer un traitementchirurgical, à type de canthopexies.
■ Pathologie cornéenneRappelons brièvement que la cornée se compose de plusieurs
couches, d’avant en arrière : l’épithélium cornéen qui est lacouche la plus superficielle ; la membrane de Bowman séparel’épithélium de la couche stromale ; le stroma cornéen estcomposé de fibres de collagène régulièrement apposées, ce quidonne sa transparence à la cornée ; la membrane de Descemet ;la couche endothéliale, dont l’intégrité est indispensable aumaintien de la transparence cornéenne, car cette coucheconstitue une barrière entre l’humeur aqueuse présente dans lachambre antérieure de l’œil et le stroma cornéen (en cas dedysfonctionnement endothélial, un œdème cornéen survient).
KératitesLes kératites sont caractérisées par une atteinte infectieuse ou
inflammatoire de la cornée. Une kératite superficielle, atteignantl’épithélium, est diagnostiquée par le test à la fluorescéine : uneérosion est alors visible en lumière bleu cobalt. Les étiologiessont multiples. La kératite herpétique a déjà été évoquée.L’atteinte rubéolique congénitale peut, quant à elle, entraînerune opacité séquellaire.
Dystrophies héréditaires de cornée [22]
La classification des dystrophies héréditaires de la cornée abénéficié des récents progrès de la génétique. La plupart desdystrophies héréditaires de cornée sont de transmission autoso-mique dominante, par atteinte du gène BIGH3, situé en 5q31 etcodant pour une protéine de 683 acides aminés : lakératoépithéline.
■ Glaucome et dysgénésiesdu segment antérieur
Nous ne pouvons commencer cette courte approche duglaucome congénital sans citer le rapport « Œil et génétique »de la Société française d’ophtalmologie 2005 : « Comme lestrabisme et le nystagmus, le glaucome congénital est à la foisune maladie et un symptôme. » [23]. En effet, le glaucomecongénital dans sa forme isolée : le glaucome congénital primitifclassique, constitue une maladie en soi. Associé à d’autresmalformations du segment antérieur, il constitue un symptôme.
Isolé ou associé, le glaucome congénital résulte d’une hyper-
tonie oculaire, grave, dont le traitement est une urgence chez letout-petit. L’hypertonie oculaire, comme chez l’adulte, mais defaçon beaucoup plus rapide, entraîne une atrophie optique parischémie progressive du nerf optique. L’autre conséquence del’hypertonie oculaire, mais ceci à la différence de l’adulte, estune opacification de la cornée par rupture de la membrane deDescemet, et une augmentation de la taille de la cornée et duglobe dans son ensemble, qui est encore élastique à cet âge.L’œdème de cornée, au départ, pouvant régresser si le tonusoculaire est très rapidement normalisé, va laisser la place à desopacifications définitives bleutées par désorganisation del’architecture collagène du stroma cornéen. C’est dire combienle traitement chirurgical du glaucome congénital est urgent(Fig. 3, 4).
Le glaucome congénital primitif classique résulterait d’undéfaut d’apoptose des cellules du trabéculum, d’où défaut derésorption de l’humeur aqueuse. Il atteindrait une naissance sur20 000. La transmission en est autosomique récessive, avecidentification du gène CYP1B1 codant le cytochrome P450 detype 1B1.
Figure 3. Glaucome congénital à 2 jours de vie. Buphtalmie bilatérale etœdème de cornée entraînant une opacification débutante des cornées.
Figure 4.A. Glaucome congénital évolué avec opacité définitive de cornée (quinécessitera une greffe de cornée).B. Opacité séquellaire de glaucome opéré avec un retard de diagnostic.
650651652653654655656657658659660661662663664665
666667668669670671672673
674
675676677678679680681682683684685
686
687688689690691692693
694
695696697698699700
701
702
703704705706707708709710
711
712
713
714
715
716
717
718
719
720
721
722
723
724
725
726
727
728
729
730
731
.
.
Ophtalmologie infantile ¶ 4-120-A-10
7Pédiatrie
DiagnosticLe diagnostic, parfois dans les tout premiers jours de vie mais
parfois plus tardif, repose sur l’association de plusieurs élé-ments : buphtalmie (augmentation du diamètre cornéen de plusde 1 mm, normalement de 9,5 mm à la naissance, de 10,5 mmà 6 mois, de 11,5 mm à 1 an, de 12 à 12,5 mm, c’est-à-dire lataille adulte, à 3 ans, et augmentation de la longueur axiale duglobe mesurée en échographie) ; œdème cornéen plus ou moinsimportant (épaisseur cornéenne normale inférieure à 650 µm,mesurée en pachymétrie) ; une augmentation de l’excavationpapillaire si le fond d’œil est accessible (aspect du nerf optiqueexcavé en cas d’hypertonie oculaire, alors que l’excavation estcotée inférieure à 0,1 dans les cas normaux) ; hypertonieoculaire mesurée au tonomètre de Perkins, tenant compte del’anesthésie générale qui minore de 30 à 40 % la valeur detonus oculaire (on estime donc qu’un tonus oculaire supérieurà 4 mmHg avant 1 an est pathologique, qu’il doit demeurersous le seuil de 10 mmHg avant 5 ans). Quoi qu’il en soit, lasuspicion d’un glaucome congénital (en consultation, onapprécie la transparence cornéenne, la buphtalmie et éventuel-lement l’excavation papillaire) doit conduire en urgence à laréalisation d’un examen sous anesthésie générale suivi le caséchéant, en cas de confirmation du diagnostic, d’une interven-tion chirurgicale filtrante uni- ou bilatérale à type de sclérecto-mie profonde.
SurveillanceUn contrôle est nécessaire au cours du second mois, sous
anesthésie, avec éventuelle reprise chirurgicale en cas de récidive(la plus souvent par fibrose du volet scléral du fait des phéno-mènes de cicatrisation exacerbés chez l’enfant) ; les contrôlessous anesthésie générale sont itératifs dans la petite enfance. Lesséquelles opaques de cornée peuvent nécessiter à terme unekératoplastie transfixiante. La correction d’une myopie axilerésultante de l’augmentation de la longueur du globe estnécessaire.
Formes cliniquesLe glaucome symptôme s’associe à une malformation du
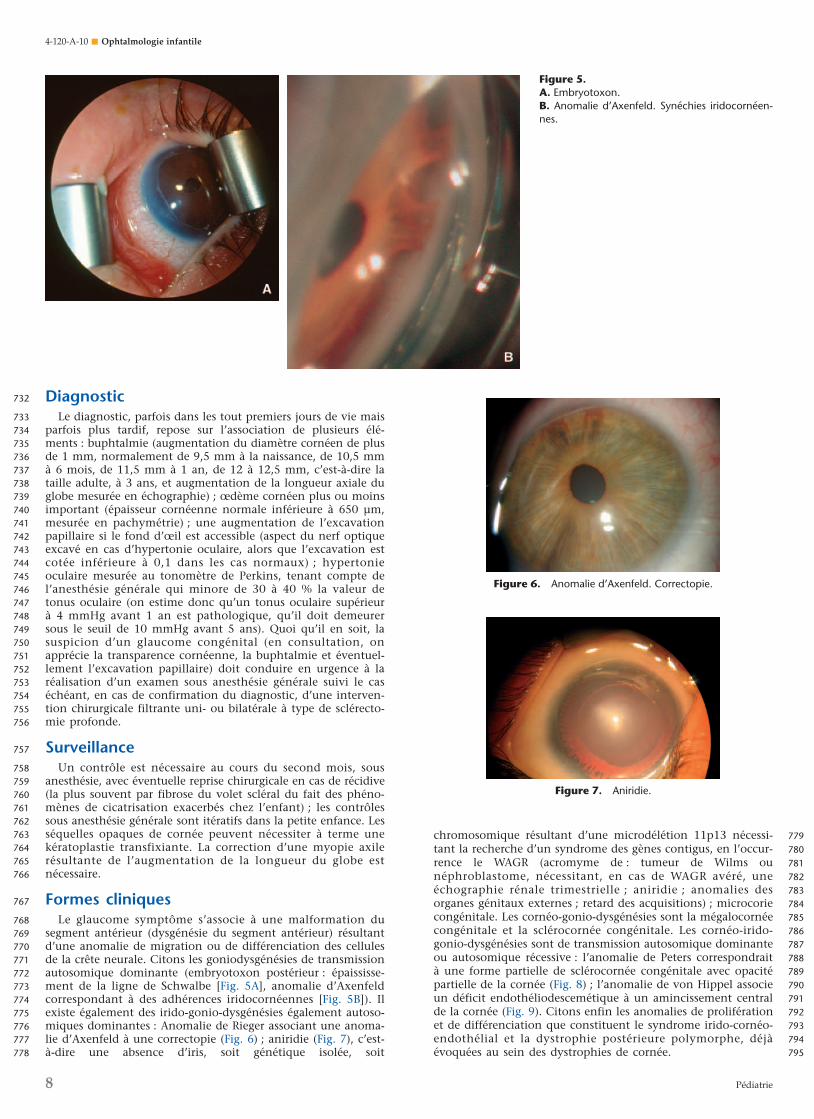
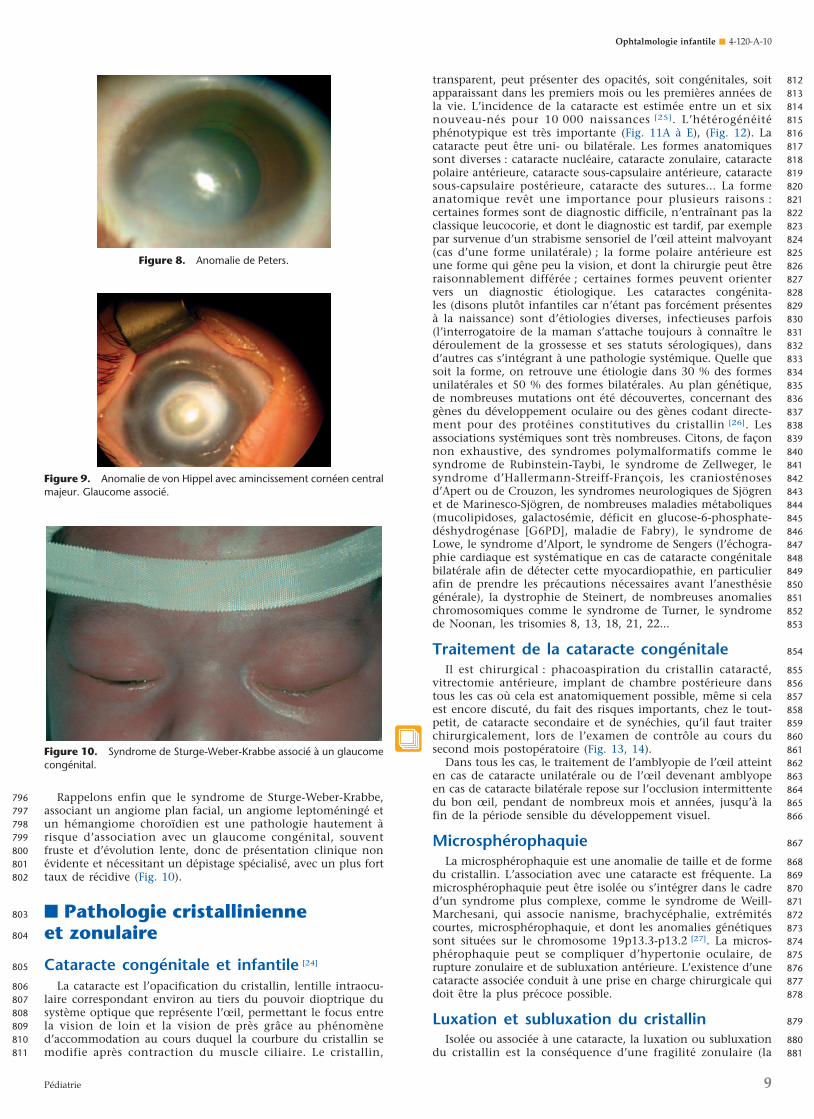
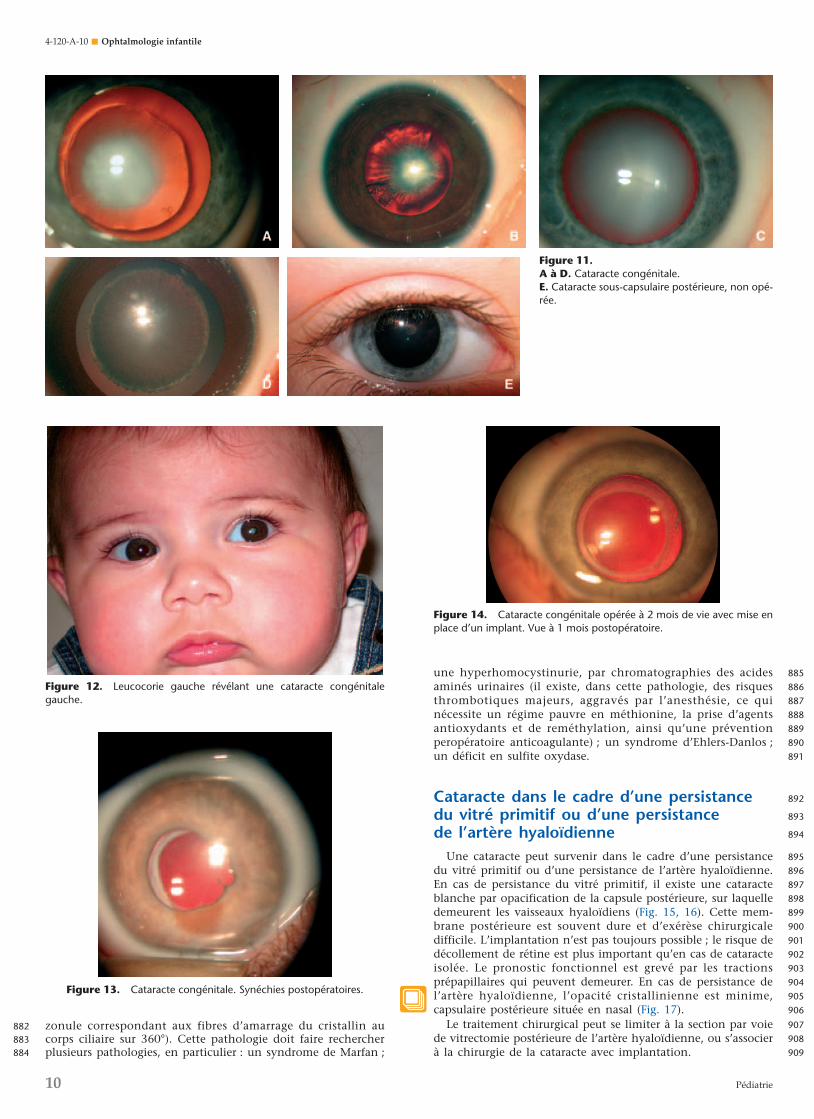
segment antérieur (dysgénésie du segment antérieur) résultantd’une anomalie de migration ou de différenciation des cellulesde la crête neurale. Citons les goniodysgénésies de transmissionautosomique dominante (embryotoxon postérieur : épaississe-ment de la ligne de Schwalbe [Fig. 5A], anomalie d’Axenfeldcorrespondant à des adhérences iridocornéennes [Fig. 5B]). Ilexiste également des irido-gonio-dysgénésies également autoso-miques dominantes : Anomalie de Rieger associant une anoma-lie d’Axenfeld à une correctopie (Fig. 6) ; aniridie (Fig. 7), c’est-à-dire une absence d’iris, soit génétique isolée, soit
chromosomique résultant d’une microdélétion 11p13 nécessi-tant la recherche d’un syndrome des gènes contigus, en l’occur-rence le WAGR (acromyme de : tumeur de Wilms ounéphroblastome, nécessitant, en cas de WAGR avéré, uneéchographie rénale trimestrielle ; aniridie ; anomalies desorganes génitaux externes ; retard des acquisitions) ; microcoriecongénitale. Les cornéo-gonio-dysgénésies sont la mégalocornéecongénitale et la sclérocornée congénitale. Les cornéo-irido-gonio-dysgénésies sont de transmission autosomique dominanteou autosomique récessive : l’anomalie de Peters correspondraità une forme partielle de sclérocornée congénitale avec opacitépartielle de la cornée (Fig. 8) ; l’anomalie de von Hippel associeun déficit endothéliodescemétique à un amincissement centralde la cornée (Fig. 9). Citons enfin les anomalies de proliférationet de différenciation que constituent le syndrome irido-cornéo-endothélial et la dystrophie postérieure polymorphe, déjàévoquées au sein des dystrophies de cornée.
Figure 5.A. Embryotoxon.B. Anomalie d’Axenfeld. Synéchies iridocornéen-nes.
Figure 6. Anomalie d’Axenfeld. Correctopie.
Figure 7. Aniridie.
732
733734735736737738739740741742743744745746747748749750751752753754755756
757
758759760761762763764765766
767
768769770771772773774775776777778
779780781782783784785786787788789790791792793794795
4-120-A-10 ¶ Ophtalmologie infantile
8 Pédiatrie
Rappelons enfin que le syndrome de Sturge-Weber-Krabbe,associant un angiome plan facial, un angiome leptoméningé etun hémangiome choroïdien est une pathologie hautement àrisque d’association avec un glaucome congénital, souventfruste et d’évolution lente, donc de présentation clinique nonévidente et nécessitant un dépistage spécialisé, avec un plus forttaux de récidive (Fig. 10).
■ Pathologie cristallinienneet zonulaire
Cataracte congénitale et infantile [24]
La cataracte est l’opacification du cristallin, lentille intraocu-laire correspondant environ au tiers du pouvoir dioptrique dusystème optique que représente l’œil, permettant le focus entrela vision de loin et la vision de près grâce au phénomèned’accommodation au cours duquel la courbure du cristallin semodifie après contraction du muscle ciliaire. Le cristallin,
transparent, peut présenter des opacités, soit congénitales, soitapparaissant dans les premiers mois ou les premières années dela vie. L’incidence de la cataracte est estimée entre un et sixnouveau-nés pour 10 000 naissances [25]. L’hétérogénéitéphénotypique est très importante (Fig. 11A à E), (Fig. 12). Lacataracte peut être uni- ou bilatérale. Les formes anatomiquessont diverses : cataracte nucléaire, cataracte zonulaire, cataractepolaire antérieure, cataracte sous-capsulaire antérieure, cataractesous-capsulaire postérieure, cataracte des sutures... La formeanatomique revêt une importance pour plusieurs raisons :certaines formes sont de diagnostic difficile, n’entraînant pas laclassique leucocorie, et dont le diagnostic est tardif, par exemplepar survenue d’un strabisme sensoriel de l’œil atteint malvoyant(cas d’une forme unilatérale) ; la forme polaire antérieure estune forme qui gêne peu la vision, et dont la chirurgie peut êtreraisonnablement différée ; certaines formes peuvent orientervers un diagnostic étiologique. Les cataractes congénita-les (disons plutôt infantiles car n’étant pas forcément présentesà la naissance) sont d’étiologies diverses, infectieuses parfois(l’interrogatoire de la maman s’attache toujours à connaître ledéroulement de la grossesse et ses statuts sérologiques), dansd’autres cas s’intégrant à une pathologie systémique. Quelle quesoit la forme, on retrouve une étiologie dans 30 % des formesunilatérales et 50 % des formes bilatérales. Au plan génétique,de nombreuses mutations ont été découvertes, concernant desgènes du développement oculaire ou des gènes codant directe-ment pour des protéines constitutives du cristallin [26]. Lesassociations systémiques sont très nombreuses. Citons, de façonnon exhaustive, des syndromes polymalformatifs comme lesyndrome de Rubinstein-Taybi, le syndrome de Zellweger, lesyndrome d’Hallermann-Streiff-François, les craniosténosesd’Apert ou de Crouzon, les syndromes neurologiques de Sjögrenet de Marinesco-Sjögren, de nombreuses maladies métaboliques(mucolipidoses, galactosémie, déficit en glucose-6-phosphate-déshydrogénase [G6PD], maladie de Fabry), le syndrome deLowe, le syndrome d’Alport, le syndrome de Sengers (l’échogra-phie cardiaque est systématique en cas de cataracte congénitalebilatérale afin de détecter cette myocardiopathie, en particulierafin de prendre les précautions nécessaires avant l’anesthésiegénérale), la dystrophie de Steinert, de nombreuses anomalieschromosomiques comme le syndrome de Turner, le syndromede Noonan, les trisomies 8, 13, 18, 21, 22...
Traitement de la cataracte congénitaleIl est chirurgical : phacoaspiration du cristallin cataracté,
vitrectomie antérieure, implant de chambre postérieure danstous les cas où cela est anatomiquement possible, même si celaest encore discuté, du fait des risques importants, chez le tout-petit, de cataracte secondaire et de synéchies, qu’il faut traiterchirurgicalement, lors de l’examen de contrôle au cours dusecond mois postopératoire (Fig. 13, 14).
Dans tous les cas, le traitement de l’amblyopie de l’œil atteinten cas de cataracte unilatérale ou de l’œil devenant amblyopeen cas de cataracte bilatérale repose sur l’occlusion intermittentedu bon œil, pendant de nombreux mois et années, jusqu’à lafin de la période sensible du développement visuel.
MicrosphérophaquieLa microsphérophaquie est une anomalie de taille et de forme
du cristallin. L’association avec une cataracte est fréquente. Lamicrosphérophaquie peut être isolée ou s’intégrer dans le cadred’un syndrome plus complexe, comme le syndrome de Weill-Marchesani, qui associe nanisme, brachycéphalie, extrémitéscourtes, microsphérophaquie, et dont les anomalies génétiquessont situées sur le chromosome 19p13.3-p13.2 [27]. La micros-phérophaquie peut se compliquer d’hypertonie oculaire, derupture zonulaire et de subluxation antérieure. L’existence d’unecataracte associée conduit à une prise en charge chirurgicale quidoit être la plus précoce possible.
Luxation et subluxation du cristallinIsolée ou associée à une cataracte, la luxation ou subluxation
du cristallin est la conséquence d’une fragilité zonulaire (la
Figure 8. Anomalie de Peters.
Figure 9. Anomalie de von Hippel avec amincissement cornéen centralmajeur. Glaucome associé.
Figure 10. Syndrome de Sturge-Weber-Krabbe associé à un glaucomecongénital.
796797798799800801802
803
804
805
806807808809810811
812813814815816817818819820821822823824825826827828829830831832833834835836837838839840841842843844845846847848849850851852853
854
855856857858859860861862863864865866
867
868869870871872873874875876877878
879
880881
.
Ophtalmologie infantile ¶ 4-120-A-10
9Pédiatrie
zonule correspondant aux fibres d’amarrage du cristallin aucorps ciliaire sur 360°). Cette pathologie doit faire rechercherplusieurs pathologies, en particulier : un syndrome de Marfan ;
une hyperhomocystinurie, par chromatographies des acidesaminés urinaires (il existe, dans cette pathologie, des risquesthrombotiques majeurs, aggravés par l’anesthésie, ce quinécessite un régime pauvre en méthionine, la prise d’agentsantioxydants et de reméthylation, ainsi qu’une préventionperopératoire anticoagulante) ; un syndrome d’Ehlers-Danlos ;un déficit en sulfite oxydase.
Cataracte dans le cadre d’une persistancedu vitré primitif ou d’une persistancede l’artère hyaloïdienne
Une cataracte peut survenir dans le cadre d’une persistancedu vitré primitif ou d’une persistance de l’artère hyaloïdienne.En cas de persistance du vitré primitif, il existe une cataracteblanche par opacification de la capsule postérieure, sur laquelledemeurent les vaisseaux hyaloïdiens (Fig. 15, 16). Cette mem-brane postérieure est souvent dure et d’exérèse chirurgicaledifficile. L’implantation n’est pas toujours possible ; le risque dedécollement de rétine est plus important qu’en cas de cataracteisolée. Le pronostic fonctionnel est grevé par les tractionsprépapillaires qui peuvent demeurer. En cas de persistance del’artère hyaloïdienne, l’opacité cristallinienne est minime,capsulaire postérieure située en nasal (Fig. 17).
Le traitement chirurgical peut se limiter à la section par voiede vitrectomie postérieure de l’artère hyaloïdienne, ou s’associerà la chirurgie de la cataracte avec implantation.
Figure 11.A à D. Cataracte congénitale.E. Cataracte sous-capsulaire postérieure, non opé-rée.
Figure 12. Leucocorie gauche révélant une cataracte congénitalegauche.
Figure 13. Cataracte congénitale. Synéchies postopératoires.
Figure 14. Cataracte congénitale opérée à 2 mois de vie avec mise enplace d’un implant. Vue à 1 mois postopératoire.
882883884
885
886
887
888
889
890
891
892
893
894
895
896
897
898
899
900
901
902
903
904
905
906
907
908
909
.
4-120-A-10 ¶ Ophtalmologie infantile
10 Pédiatrie

■ Pathologie rétinienneRétinopathie des prématurés
Il s’agit d’une pathologie sévère dont la forme la plus évoluée,la fibroplasie rétrolentale, est cécitante.
La prévention et le dépistage de la rétinopathie des prématu-rés sont fondamentaux, même si la prise en charge restedifficile [28]. La physiopathologie est directement liée à l’oxygé-nothérapie [29, 30], ce qui a permis d’établir des recommanda-tions dans la prie en charge des nouveau-nés prématurés, avecen particulier un monitorage précis de la saturation en oxygène,afin que celle-ci ne dépasse pas 95 % de saturation du sang enO2
[31]. Ainsi, la fréquence de la pathologie a pu diminuer, demême que les formes les plus sévères, grâce au dépistageophtalmologique selon des procédures standardisées chez lesnouveau-nés à risque [32-34].
Outre l’hyperoxie, d’autres facteurs de risque sont connus :petit poids de naissance (inférieur à 1 500 g), terme de naissance(inférieur à 32 semaines d’aménorrhée), ventilation assistée,transfusions sanguines et traitement par surfactant pulmonaire.Il semble exister des facteurs protecteurs : traitement corticoïdepré-partum améliorant la maturation pulmonaire, vitaminothé-rapie E, photothérapie. La rétinopathie des prématurés est la
conséquence d’un trouble du développement de la vascularisa-tion rétinienne. Normalement, celle-ci se constitue depuis lapapille, du centre vers la périphérie, de la 16e à la 36e semainede vie intra-utérine. Chez le prématuré, alors que la vasculari-sation rétinienne se poursuit, l’oxygénothérapie induit unevasoconstriction capillaire et un arrêt de la progression vascu-laire. En aval de la zone d’arrêt, une zone d’ischémie se crée ;la rétine avasculaire hypoxique sécrète des facteurs angiogéni-ques. C’est l’angiogenèse pathologique qui entraîne les anoma-lies rétinovitréennes responsables in fine du décollement derétine [35].
La classification en cinq stades de la rétinopathie du préma-turé repose sur les travaux de Hindle et al. en 1984 et 1987 [36-
38]. La rétine est divisée en trois zones et 12 quadrants horaires.La zone un est un disque centré sur la papille, dont le rayon estégal à deux distances inter-papillo-maculaires. La zone deuxreprésente une couronne centrée sur la papille, excluant la zoneun, et dont le rayon est égal à la distance entre le centre de lapapille et l’ora serrata nasale. La zone trois correspond aucroissant rétinien temporal résiduel. Douze quadrants de 30°d’arc sont définis.
La rétinopathie est classée en différents stades :• le stade 0 correspond à l’absence de signe de rétinopathie ;• au stade 1, il existe une ligne de démarcation entre la rétine
normale et la zone avasculaire périphérique grisâtre ;• au stade 2, il existe une bande de démarcation large et
saillante entre la rétine normale et la zone avasculairepériphérique. La prolifération fibrovasculaire, sous la forme depetits bouquets de néovaisseaux en arrière de la bande dedémarcation, demeure dans le plan rétinien ;
• au stade 3, on observe un bourrelet saillant, surélevé parrapport au plan rétinien car la prolifération fibrovasculaire setrouve dans le vitré (Fig. 18). Il peut exister des hémorragies.Une photocoagulation au laser des zones ischémiques peutêtre nécessaire ; l’alternative est une cryothérapie élective deces mêmes zones ;
• le stade 4 correspond à un décollement de rétine tractionnel,soit avec respect maculaire, soit avec atteinte maculaire, àtype de pli falciforme (Fig. 19). Le traitement associe uncerclage chirurgical, à enlever à 3 mois, à une destruction deszones d’ischémie, soit au laser, soit par cryothérapie ;
• au stade 5, la rétine est totalement décollée, en « tunnel » ou« parapluie » : c’est l’évolution ultime vers la fibroplasierétrolentale, cécitante avec son cortège de cataracte, d’hyper-tonie oculaire et d’opacification de cornée (Fig. 20, 21). Lachirurgie à type de phacophagie, dissection de la fibroseprérétinienne, pexie, tamponnement interne peut être tentéé,même si le pronostic fonctionnel est très réservé ;
• le stade « plus » correspond à la présence de dilatationsveineuses et de tortuosités artérioveineuses, témoignant d’uneincompétence vasculaire, facteur de risque d’évolutivité de lamaladie. La rétinopathie du prématuré ne débute qu’à partirde la 4e ou 5e semaine de vie. Elle peut évoluer alors mêmeque les facteurs de risque comme l’hyperoxie ont depuislongtemps disparu.
Rétinopathies pigmentaires [39]
Les rétinopathies pigmentaires constituent un groupe depathologies d’une hétérogénéité phénotypique majeure, qu’estvenue confirmer la génétique, montrant une hétérogénéitégénotypique tout aussi prégnante. Il s’agit – le raccourci estcertes audacieux – de pathologies du métabolisme des photoré-cepteurs, touchant 1,5 million de patients dans le monde.L’amaurose congénitale de Leber, de transmission autosomiquerécessive, constitue la forme la plus sévère de ce groupe depathologies. L’enfant atteint n’exprime pas d’éveil visuel, nesourit pas au contact d’un visage ; on retrouve le signe del’éventail (l’enfant passe ses mains devant le visage), unnystagmus de malvoyance, le signe digito-oculaire de Frances-chetti. L’examen du fond d’œil ne retrouve au départ qu’unegracilité des vaisseaux, et la présence d’« ostéoblastes », c’est-à-dire des pigmentations noires diffuses commençant en périphé-rie, ne se manifeste que plus tardivement. L’électro-
Figure 15. Persistance du vitré primitif.
Figure 16. Persistance du vitré primitif évoluée avec cristallin repoussécontre la face postérieure de la cornée opacifiée.
Figure 17. Persistance de l’artère hyaloïdienne.
910
911
912913914915916917918919920921922923924925926927928929930931
932933934935936937938939940941942
943944945946947948949950951952953954955956957958959960961962963964965966967968969970971972973974975976977978979980981982983984985986
987
9889899909919929939949959969979989991000100110021003
Ophtalmologie infantile ¶ 4-120-A-10
11Pédiatrie

rétinogramme est l’examen capital : il retrouve une absence deréponse à la fois en stimulation scotopique (étude du systèmedes bâtonnets) et en stimulation photopique (étude du systèmedes cônes).
La rétinopathie pigmentaire précoce constitue une formemoins sévère d’apparition retardée entre 6 mois et 2 ans.
Les formes plus tardives encore débutent par une gêne à lavision nocturne, avec évolution progressive vers un rétrécisse-ment concentrique du champ visuel jusqu’à l’atteinte de lavision centrale ; retenons que les forme débutant avant 10 anssont plutôt de transmission récessive liées à l’X et que lesformes plus tardives débutant après l’âge de 20 ans sont plutôtautosomiques dominantes. L’électrorétinogramme peut éven-tuellement différencier, en fonction de l’atteinte prédominantedu système des cônes ou du système des bâtonnets, unerétinopathie pigmentaire de type plutôt « cone-rod » ou « rod-cone dystrophy ».
Quoi qu’il en soit, l’analyse précise du phénotype est unenécessité, pour orienter la recherche et le conseil génétique.
Il n’existe pas à l’heure actuelle de traitement ayant prouvéson efficacité chez l’homme dans les rétinopathies pigmentaires.La protection contre les ultraviolets (UV) est recommandée. Laprise en charge d’un enfant dans une structure adaptée à sescapacités visuelles est fondamentale.
Rappelons enfin l’association avec des pathologies systémi-ques : syndrome d’Usher, syndrome de Bardet-Biedl, syndromede Senior-Locken, syndrome de Cockayne...
Rappelons enfin ici l’intérêt fondamental de l’électrophysio-logie, qui permets de faire le diagnostic de rétinopathie pig-mentaire. Nous présentons le cas clinique d’une jeune fille de12 ans, se plaignant de baisse d’acuité visuelle, chiffrée à 2 à3/10e. L’examen clinique, en particulier du fond d’œil, estnormal (les ostéoblastes sont inconstants et la gracilité vascu-laire parfois difficile à apprécier). Son champ visuel tubulairepouvait en imposer pour une rétinopathie pigmentaire (Fig. 22).Le bilan électrophysiologique redresse le diagnostic (Fig. 23, 24).Il faut rassurer l’enfant sur son trouble passager (psychosomati-que ?). De fait, sa vision s’est normalisée en quelques semaines.
Pathologies vasculaires
Maladie de Coats [40]
L’angiomatose de Leber-Coats est une affection unilatéraleentraînant des télangiectasies périphériques se compliquant dezones d’exsudation jaunâtre sous-rétinienne et de zones d’isché-mie rétinienne. Le traitement est la photocoagulation laser ou
Figure 18. Rétinopathie du prématuré stade 3 plus. Aspect de tortuo-sité vasculaire.
Figure 19. Rétinopathie du prématuré stade 4. Décollement de rétinetractionnel.
Figure 20. Rétinopathie du prématuré stade 5. Aspect échographique.
Figure 21. Rétinopathie du prématuré stade 5. Décollement de rétinetotal (fibroplasie rétrolentale).
100410051006100710081009
10101011101210131014101510161017101810191020102110221023102410251026102710281029103010311032103310341035103610371038103910401041
1042
1043
1044104510461047
4-120-A-10 ¶ Ophtalmologie infantile
12 Pédiatrie

la cryothérapie des zones d’ischémie. Dans les formes trèssévères, le décollement de rétine exsudatif peut aboutir à laperte fonctionnelle de l’œil.
Angiomatose de von Hippel-LindauIl s’agit d’une angiomatose rétinienne associée à des héman-
giomes cérébelleux et rénaux, qu’il est nécessaire de rechercher.Il s’agit d’une pathologie le plus souvent sporadique, mais danscertains cas autosomique dominante. Les hémangiomes réti-niens peuvent se compliquer d’exsudats maculaires
et de décollement de rétine exsudatif. Le traitement deshémangiomes rétiniens consiste en une photocoagulation aulaser.
DrépanocytoseLa drépanocytose peut se traduire par des zones d’ischémie
rétinienne périphérique qu’il convient de photocoaguler aulaser.
Dysplasies rétinovitréennesLes dystrophies vitréorétiniennes héréditaires sont des
pathologies rares, au pronostic visuel réservé du fait du risquede décollement de rétine. Ces pathologies sont nombreuses, auxphénotypes variables en fonction de l’expressivité des signes
cliniques [41-43]. Ces dégénérescences vitréorétiniennes représen-tent un ensemble variable de pathologies, dont la physiopatho-logie de chacune d’entre elles reste mal connue, malgré lesrécentes découvertes génétiques associant certaines de cespathologies à des anomalies de gènes codant pour le collagène.Ces dystrophies vitréorétiniennes peuvent s’associer à unemyopie forte et à des manifestations systémiques.
La nosologie est imprécise, les frontières floues entre denombreux syndromes. On retrouve : le syndrome de Wagner [44-
46], le syndrome de Stickler [47], la maladie de Goldmann-Favre [48], la vitréorétinopathie exsudative familiale [49], lessyndromes de Spranger-Wiedemann et de Kniest [50], de Mars-hall [51, 52], de Weissenbacher-Zweymuller [53]. De nombreusesautres entités sont décrites.
De nombreuses autres maladies secondaires à des anomaliesdu collagène peuvent associer, mais de façon moins fréquente,une dystrophie vitréorétinienne avec ou sans myopie forte.Citons la maladie de Marfan.
À part se place le rétinoschisis congénital lié à l’X, de par samoindre rareté, son mode de transmission et sa physiopatholo-gie [54, 55].
Parmi les dystrophies vitréorétiniennes héréditaires, l’arthro-ophtalmopathie héréditaire progressive, décrite en 1965 parStickler, est une vitréorétinopathie héréditaire associant dessignes oculaires à un syndrome de Pierre Robin plus ou moinscomplet, à des atteintes osseuses et à une atteinte auditive [56,
57]. Du point de vue ophtalmologique, elle est redoutable du faitdu risque de décollement de rétine.
Les manifestations ophtalmologiques du syndrome de Sticklersont une myopie forte, une dégénérescence vitréorétiniennesévère avec brides de traction, anomalies pigmentaires, palissa-des, déchirures, décollement de rétine, cataracte nucléaire etsous-capsulaire postérieure, dysgénésie du segment antérieur,parfois strabisme, buphtalmie, glaucome.
Les manifestations générales comprennent un syndrome dePierre Robin (malformation congénitale associant : fentepalatine, glossoptôse, rétrognathie, hypoplasie mandibulaire),une surdité neurosensorielle, des anomalies squelettiques(morphotype marfanoïde), une hyperlaxité ligamentaire, unedysplasie spondyloépiphysaire.
D’autres signes sont occasionnels : cyphose, scoliose, hypoto-nie, palais court, membres longs, anomalie de l’émail, anomaliedes diaphyses, anodontie ou oligodontie, mauvais articulédentaire, amyotrophie, agénésie de muscles, rétrognathisme,micrognathisme, mains longues, arachnodactylie, maigreur
Figure 22. Rétinopathie pigmentaire. Cas clinique. Champ visueltubulaire.
Figure 23. Rétinopathie pigmentaire. Cas clinique. Électrorétinogramme normal.
104810491050
1051
10521053105410551056105710581059
1060
106110621063
1064
1065106610671068
1069107010711072107310741075
1076107710781079108010811082
1083108410851086
108710881089
1090109110921093109410951096
109710981099110011011102
110311041105110611071108
11091110111111121113
.
.
Ophtalmologie infantile ¶ 4-120-A-10
13Pédiatrie

(autre que lipodystrophie), pommettes plates par hypoplasie desmalaires, éruption précoce des dents, présence de dentsnéonatales.
L’anomalie en cause est une mutation du collagène, avechétérogénéité au point de vue génétique [58-60].
L’expressivité de l’affection est très variable ; le syndrome deWagner serait une forme de la maladie atteignant uniquementl’œil, sans manifestations rhumatologiques, et avec une atteintemoins sévère (moindre fréquence de décollement de rétine).Dans ce syndrome, les mutations du gène du collagène II sonten effet retrouvées au niveau de l’exon 2 du collagène IIA1, etcet exon n’est pas transcrit dans les tissus extraoculaires [61].
Le risque de décollement de rétine est majeur dans lesdystrophies vitréorétiniennes ; en conséquence, plusieursthérapeutiques préventives ont été proposées [62, 63] : cerclagechirurgical avec indentation, qui semble être la seule thérapeu-tique efficace, le barrage laser à l’argon pouvant au contraireêtre délétère pour certains auteurs : il induit une cicatrice descouches profondes de la rétine, alors que les tissus pathologi-ques sont au niveau des couches internes de la rétine et del’interface vitréorétinienne. En l’absence de facteurs pronosti-ques précis, les indications de ces différentes thérapeutiquespréventives sont mal codifiées.
Le décollement de rétine dans les dystrophies vitréorétinien-nes est de mécanisme rhegmatogène : dans le rétinoschisis, bienconnu, le mécanisme de décollement de rétine est attribué à latransformation d’un trou dans le mur interne de la rétine,constitué après plusieurs années d’évolution de la bulle deschisis à la paroi fine et fragile, en un trou complet associant untrou du mur externe ; ceci constitue la déchirure qui, associéeaux tractions vitréennes d’un vitré pathologique, va entraîner ledécollement de rétine.
Dyschromatopsies héréditaires [64]
Le diagnostic d’un trouble de la vision des couleurs peut êtreimportant, en particulier pour l’orientation professionnellefuture. Les dyschromatopsies héréditaires résultent d’uneanomalie d’un ou plusieurs cônes (qui sont des photorécepteursrétiniens), par absence d’un photopigment ou modification descaractéristiques d’absorption spectrale du pigment, c’est-à-direvariation du maximum de sensibilité du pigment. Il existe troistypes de cône et chacun possède un pigment qui a un maxi-mum de sensibilité à la lumière correspondant à une longueurd’onde précise. Les cônes L ont un maximum de sensibilité à560 nm de longueur d’onde. Ce sont les cônes « rouges ». Lescônes M ont un maximum de sensibilité à 530 nm de longueur
d’onde. Ce sont les cônes « verts ». Les cônes S ont un maxi-mum de sensibilité à 420 nm de longueur d’onde. Ce sont lescônes « bleus ». La vision des couleurs est donc trichromati-que (théorie de Young-Helmholtz). L’analyse du signal lumineuxest réalisée dès l’étage des cellules ganglionnaires rétiniennesselon des antagonismes rouge/vert et bleu/jaune (rouge + vert).C’est la théorie de Hering.
La classification des dyschromatopsies regroupe d’abord letrichromatisme anormal, dans lequel le sujet possède les troistypes de cônes mais avec une anomalie d’un photopigment. Laprotanomalie est liée à une atteinte des cônes L ; elle estautosomique récessive liée à l’X, et atteindrait en Europe 1 %des garçons et 0,02 % des filles. La deutéranomalie est liée à uneatteinte des cônes M ; elle est autosomique récessive liée à l’X,et atteindrait en Europe 5 % des garçons et 0,04 % des filles. Latritanomalie est liée à une atteinte des cônes S ; elle estautosomique dominante à pénétrance variable, liée au chromo-some 7. Dans le dichromatisme anormal, il y a absence defonctionnement d’un photopigment. C’est le véritable « dalto-nisme ». La protanopie est liée à l’absence de cône L ; elle estrécessive liée à l’X et atteindrait 1 % des garçons et 0,02 % desfilles. La deutéranopie est liée à l’absence de cône M ; elle estrécessive liée à l’X et atteindrait 1,4 % des garçons et 0,01 % desfilles. La tritanopie est liée à l’absence de cône S ; elle estautosomique dominante liée au chromosome 7. Le monochro-matisme à cônes bleus est une pathologie au cours de laquelleseuls les cônes de type S fonctionnent. C’est une pathologierécessive liée à l’X ; il peut exister une baisse d’acuité visuelle,un nystagmus. Dans l’achromatopsie, seuls les bâtonnets (quisont le second type de photorécepteur) sont fonctionnels ; c’estune pathologie autosomique récessive ; l’acuité visuelle estbasse, il existe une photophobie, parfois un nystagmus bilatéralde malvoyance. C’est l’électrorétinogramme qui fait le diagnos-tic différentiel entre ces deux dernières pathologies : dans lemonochromatisme à cônes bleus, l’électrorétinogramme « cone »est discernable mais d’amplitude diminuée (il teste les troistypes de cônes) et l’électrorétinogramme « flicker » n’est pasdiscernable (il ne teste que les cônes L et M) ; dans l’achroma-topsie, ni l’électrorétinogramme « cone » ni l’électrorétino-gramme « flicker » n’est discernable.
Maladie de Stargardt ou fundusflavimaculatus
Il s’agit d’une maculopathie de transmission autosomiquerécessive, survenant dans la seconde enfance. La vision chute enquelques années pour se stabiliser entre 1/20e et 2/10e environ.
Figure 24. Rétinopathie pigmentaire. Cas clinique. Potentiels évoqués visuels normaux.
11141115111611171118111911201121112211231124112511261127112811291130113111321133113411351136113711381139114011411142114311441145
1146
114711481149115011511152115311541155115611571158
1159116011611162116311641165116611671168116911701171117211731174117511761177117811791180118111821183118411851186118711881189119011911192119311941195119611971198
1199
1200
120112021203
.
.
4-120-A-10 ¶ Ophtalmologie infantile
14 Pédiatrie

L’examen du fond d’œil, normal au début de la maladie,retrouve des altérations bilatérales : aspect de la macula jaunâtreavec reflets en « bronze martelé » au début de la maladie,évoluant avec les années vers des remaniements atrophiquesavec migrations pigmentaires. La zone périmaculaire est le siègede taches blanchâtres correspondant au fundus flavimaculatus.La génétique de cette maladie repose sur des mutations variablesdu gène ABCA4 en 1p22.
Malformations rétiniennes et du nerfoptique : « morning glory syndrome »,colobomes du nerf optique et de la rétine,fossette colobomateuse
La pathologie malformative du nerf optique prend, quant àelle, des aspects variables plus ou moins sévères, depuis lemorning glory syndrome (Fig. 25, 26)
malheureusement incompatible avec une fonction visuellenormale, à la fossette colobomateuse, qui peut rester asympto-matique (Fig. 27). En cas de décollement séreux rétinien, letraitement associe une photocoagulation péripapillaire à uneinjection de gaz en intravitréen avec positionnement postopé-ratoire (ce qui peut faire préférer chez l’enfant une vitrectomieavec tamponnement complet de la cavité oculaire). Le colo-bome rétinien et du nerf optique entraîne une amputation duchamp visuel, mais peut laisser une acuité visuelle normale enl’absence d’atteinte maculaire.
■ StrabismesNous avons déjà largement évoqué dans ce traité la patholo-
gie strabique de l’enfant [65]. Nous rappellerons qu’un strabismen’est jamais normal ; il peut résulter d’un trouble oculomoteurpropre, ou révéler une pathologie organique sous-jacente
(puisqu’un œil qui ne voit pas ne peut pas fixer). Quoi qu’il ensoit, le diagnostic, ou ne serait-ce qu’un doute sur un strabismedans les premiers mois de vie, requiert un examen spécialiséavec un fond d’œil. Si l’examen anatomique est normal, alorsc’est l’évolution de la déviation qui signera ou pas le réeldiagnostic de strabisme, nécessitant alors une prise en chargespécifique. Encore une fois, le rétinoblastome, pathologieexceptionnelle, peut tout de même se révéler par une déviationoculaire. C’est dire la nécessité de l’examen attentif du fondd’œil en cas de strabisme.
Une déviation strabique permanente est une hétérotropie. Ondistingue les ésotropies (strabismes convergents) des exotropies(strabismes divergents). Une déviation strabique latente est unehétérophorie. On distingue les ésophories des exophories. Lesstrabismes verticaux sont décrits relativement à l’œil dévié versle haut : on parle d’hyperphorie ou d’hypertropie. Les strabis-mes en torsion sont consécutifs à des troubles des mouvementsde cyclotorsion : on parle de cyclotropie ou cyclophorie. Unépicanthus donne un faux aspect de strabisme : les refletscornéens sont centrés (Fig. 28).
Nous rappellerons brièvement les différents types de stra-bisme de l’enfant.• Le strabisme précoce constitue une entité strabologique à
part [66], du fait d’un pronostic fonctionnel réservé en termesde qualité de la vision binoculaire espérée [67]. Le terme desyndrome de strabisme précoce est préférable à celui destrabisme congénital, car il peut survenir plusieurs mois aprèsla naissance, le plus souvent dans les 6 premiers mois. Sonorigine est mal connue mais intéresse un trouble du dévelop-pement des voies visuelles supranucléaires. Il s’agit dansl’immense majorité des cas d’une ésotropie à grand anglesupérieur à 40 dioptries (Fis. 29, 30). Il faut rechercher unealternance de fixation qui est le meilleur moyen de prévenirl’amblyopie (monoculaire). Le traitement associe bien sûr larééducation et/ou la prévention de l’amblyopie, et un traite-ment chirurgical éventuel.
• En cas de strabisme accommodatif, il existe le plus souventune hypermétropie qui entraîne une accommodation exces-sive, afin de focaliser le rayon incident sur la rétine, et demaintenir une image nette [68]. Cette accommodation suscite
Figure 25. Morning glory syndrome. Vue du fond d’œil.
Figure 26. Morning glory syndrome. Vue scanographique : aspect dekyste de la tête du nerf optique droit.
Figure 27. Fossette colobomateuse de la papille compliquée d’undécollement séreux de la rétine.
Figure 28. Épicanthus chez un enfant asiatique. Absence de strabisme.
12041205120612071208120912101211
1212
1213
1214
1215
1216121712181219122012211222122312241225122612271228
1229
1230123112321233
1234123512361237123812391240124112421243124412451246124712481249125012511252125312541255125612571258125912601261126212631264126512661267126812691270127112721273
.
Ophtalmologie infantile ¶ 4-120-A-10
15Pédiatrie

une convergence du fait du réflexe d’accommodation-convergence, soit normale, soit excessive d’où l’apparitiond’un angle de déviation. La correction de l’hypermétropiecorrige partiellement ou totalement la déviation, en soula-geant l’effort accommodatif. Il est toujours nécessaire decorriger totalement l’hypermétropie (évaluée après cycloplé-gie) car l’immense majorité des strabismes comporte une partaccommodative. Il n’existe pas d’indication chirurgicalequand le strabisme est bien corrigé par la correction optique.
• Un strabisme sensoriel ou secondaire est une déviationoculaire d’un œil amblyope (par exemple après cataractecongénitale opérée) ; la déviation est plus volontiers enconvergence dans l’enfance et e divergence à l’âge adulte(Fig. 31). Le traitement est d’abord celui de l’amblyopie, etsecondairement d’un angle résiduel le cas échéant.
• L’ésotropie tardive non accommodative est une déviation enconvergence qui survient progressivement, de façon tardivec’est-à-dire après l’âge de 3 ou 4 ans. Ce type de strabisme estpeu amblyogène car les voies visuelles et l’architecturecorticale et calleuse sont pratiquement totalement mises en
place à cet âge. Le traitement est essentiellement chirurgical,pouvant faire suite à un traitement prismatique transitoire.Les résultats sont en général bons.
• Les exotropies intermittentes ou exophories-tropies oustrabismes divergents intermittents sont de présentation assezhétérogène selon les patients [69]. Du fait de son caractèreintermittent et de sa survenue relativement tardive, elle estpeu amblyogène. La déviation est variable ; elle se manifesteplutôt en vision de loin, l’angle étant corrigé en vision deprès par la convergence accommodative mais aussi la conver-gence fusionnelle et la convergence volontaire (Fig. 32). Ilexiste donc une incomitance loin/près, avec un angle trèsvariable. L’exophorie intermittente s’aggrave le plus souventavec l’âge surtout si la déviation est supérieure à 20 dioptries,du fait de la baisse de la vergence tonique, de la diminutiondu pouvoir accommodatif et de la divergence accrue desorbites. Le traitement ne repose sur la rééducation orthopti-que qu’uniquement dans les cas de strabisme divergent parinsuffisance de convergence, c’est-à-dire lorsque la déviationest plus importante de près que de loin (situation qui n’estpas la plus fréquente). Le traitement est chirurgical dans lesautres cas mais n’empêche pas toujours une récidive pouvantnécessiter une réintervention.
• La paralysie de l’oblique supérieur est relativement fréquente.Il s’agit le plus souvent d’une atteinte de la quatrième pairecrânienne. Elle peut être unilatérale, le plus souvent congéni-tale d’étiologie inconnue ou traumatique, ou bilatérale,traumatique le plus souvent (Fig. 33). Dans la paralysiecongénitale du IV, l’enfant va présenter, dès atteint l’âge duport de la tête, un torticolis permanent et progressif ; la têteest penchée du côté opposé à l’œil atteint.
• Le syndrome de Brown est une limitation de l’action del’oblique supérieur, le plus souvent congénitale ; il estsecondaire à l’inextensibilité de l’oblique supérieur. Cecientraîne une limitation de la motilité dans le champ d’actiondu muscle oblique supérieur, et donc une impotence d’éléva-tion en adduction. Le traitement chirurgical est réservé auxatteintes sévères avec décompensation en position primaire(position des yeux fixant droit devant).
Figure 29. Strabisme convergent précoce, élévation en adductionbilatérale.
Figure 30. Strabisme convergent. Œil gauche fixateur.
Figure 31. Strabisme divergent de l’œil gauche dû à une cataractecongénitale.
Figure 32. Strabisme divergent. Vue préopératoire.
Figure 33. Paralysie bilatérale des obliques supérieurs.
12741275127612771278127912801281128212831284128512861287128812891290129112921293
129412951296129712981299130013011302130313041305130613071308130913101311131213131314131513161317131813191320132113221323132413251326132713281329133013311332
.
4-120-A-10 ¶ Ophtalmologie infantile
16 Pédiatrie

• Il existe, dans le syndrome de Stilling-Türk-Duane, unelimitation de certains mouvements oculaires, d’étiologie malconnue, attribuée à des anomalies innervationnelles, avecfibrose musculaire secondaire.Le traitement des syndromes de Stilling-Türk-Duane est
difficile et spécifique. La chirurgie peut être indiquée en cas detorticolis associé.• Les syndromes alphabétiques peuvent s’associer à toutes
formes de strabismes et entraînent une incomitance de ladéviation entre le regard en bas et le regard en haut. Ilsseraient dus à des défauts de position des insertions muscu-laires.
• Si les paralysies congénitales du III représentent la moitié desatteintes du III chez l’enfant, il ne faut bien entendu pasméconnaître les étiologies tumorales, vasculaires ou inflam-matoires. Il existe un ptosis qui peut masquer la diplopie etune déviation variable plutôt en divergence. Le traitementdépend de la cause. Le traitement de la déviation, quant à lui,peut employer des prismes, et une chirurgie n’est envisagéequ’au stade de séquelles définitives. Le vrai problème estl’amblyopie, possible en cas d’atteinte totale et précoce.
• Les causes de paralysie du VI sont multiples : causes tumorale,vasculaire, infectieuse ou inflammatoire, traumatique. Danscertains cas, la paralysie est congénitale ou d’apparitionprécoce idiopathique. Il existe une ésotropie marquée, unediplopie selon l’âge d’apparition. Le traitement est d’abordétiologique ; le traitement symptomatique associe des prismesà un traitement chirurgical des séquelles. Une occlusionalternante intermittente peut prévenir l’apparition d’uneamblyopie.
• La myasthénie peut se révéler par une atteinte oculomotricevariable, entraînant une diplopie intermittente variable.
• L’ophtalmoplégie externe progressive est une maladie rarecaractérisée par une paralysie de l’élévation survenant defaçon progressive chez l’enfant ou l’adulte jeune, s’associantà un ptosis. Une ophtalmoplégie totale s’installe progressive-ment, l’atteinte pouvant rester isolée ou s’associer à unsyndrome pseudobulbaire.
• Les craniosténoses peuvent s’accompagner de strabisme dufait d’une malposition orbitaire. La priorité dans ces hypopla-sies massives du squelette facial est la surveillance de lacornée du fait du risque majeur de kératites d’exposition, etdu fond d’œil en raison du risque d’atteinte du nerf optique.
■ Neuro-ophtalmologie
Retard de maturation visuelle ou syndromede Beauvieux
Le tableau de retard de maturation visuelle correspond à unenfant dont l’éveil visuel est pathologique dans les premiersmois de la vie, le contexte clinique pouvant évoquer uneamaurose congénitale de Leber. L’examen anatomique estnormal, si ce n’est une relative pâleur papillaire bilatérale. Lebilan électrophysiologique est fondamental : il retrouve uneélectrogenèse rétinienne (électrorétinogramme) parfaitementnormale et symétrique, alors que les PEV sont de latencediscrètement retardée pour l’âge. Il s’agit d’un retard demyélinisation des voies optiques. L’évolution est spontanémentfavorable, avec rattrapage après l’âge de 6 mois d’un comporte-ment visuel normal et un examen des PEV se normalisant.
NystagmusLe nystagmus est à la fois une maladie et un symptôme. Il est
un symptôme dans toutes les causes de malvoyance (rétinopa-thie pigmentaire, amblyopie profonde, achromatopsie congéni-tale, aniridie avec hypoplasie fovéolaire). Il est une maladieisolée, par défaut du réflexe de fixation oculaire, dans lenystagmus congénital manifeste latent ou dans le nystagmuscongénital patent. Il peut alors s’associer à un strabisme précoceet constituer une tropie nystagmique. La présence d’un nystag-mus isolé chez un enfant justifie la réalisation d’un bilan
électrophysiologique pour éliminer une neuropathie ou unerétinopathie. Le caractère atypique du nystagmus, l’associationà un examen neuropédiatrique anormal justifient la réalisationd’une imagerie cérébrale par imagerie par résonance magnétique(IRM) (Fig. 34).
Neuropathies optiques• Le glaucome congénital constitue une neuropathie optique
sévère, nous l’avons vu.• Les neuropathies optiques inflammatoires sont, chez l’enfant,
représentées par la neuropathie optique démyélinisante dansle cadre d’une sclérose en plaques, rare mais possible chezl’enfant. Il existe une baisse d’acuité visuelle variable, unediminution du réflexe photomoteur direct, une classiquedouleur à la mobilisation des globes. L’examen des PEV peutmontrer un retard des temps de culmination. Le champvisuel peut retrouver un scotome central. L’examen de lafonction de sensibilité aux contrastes retrouve une altérationde cette fonction dans les basses fréquences spatiales. Il fautrechercher à l’examen IRM des signes à la fois de dissémina-tion spatiale de la maladie (plaques multiples) et des signesde dissémination temporelle (plaques d’âges différents).
• La neuromyélite optique de Devic associe une neuropathieoptique bilatérale à une myélite transverse entraînant uneparaparésie, voire une paraplégie sensitivomotrice. Leshypersignaux en IRM sont présents au niveau des nerfsoptiques et de la moelle épinière.
• Les neuropathies optiques peuvent également entrer dans lecadre d’une pathologie inflammatoire systémique comme lasarcoïdose, d’une pathologie infectieuse (herpès, tuberculose,toxoplasmose), d’une étiologie toxique (éthambutol,monoxyde de carbone). La neuropathie optique peut êtrecompressive, et un œdème papillaire uni- ou bilatéral, voireunilatéral avec atrophie optique controlatérale réalisant lesyndrome de Foster-Kennedy doit faire rechercher unetumeur cérébrale et/ou au contact des voies optiques.
• Le gliome du nerf optique de l’enfant est un gliome histolo-giquement « bénin » à la différence du gliome malin del’adulte. Il s’agit d’un astrocytome fibrillaire ou pilocytique àpotentiel évolutif faible : il n’y a pas d’infiltration du nerfoptique, mais une compression possible. À l’IRM, la lésion estfusiforme aux limites nettes. Dans un tiers des cas, il s’associeà une maladie de Recklinghausen (neurofibromatose type I).Une surveillance clinique et radiologique s’impose. La
Figure 34. Lésion démyélinisante du tronc cérébral révélée par unstrabisme vertical d’apparition brutale avec nystagmus rotatoire.
1333133413351336133713381339134013411342134313441345134613471348134913501351135213531354135513561357135813591360136113621363136413651366136713681369137013711372137313741375
1376
1377
1378
137913801381138213831384138513861387138813891390
1391
139213931394139513961397139813991400
14011402140314041405
1406
1407140814091410141114121413141414151416141714181419142014211422142314241425142614271428142914301431143214331434143514361437143814391440144114421443
.
Ophtalmologie infantile ¶ 4-120-A-10
17Pédiatrie
chirurgie ne se justifie qu’en cas de baisse de vision, d’altéra-tion franche du champ visuel et/ou des PEV. Rappelons quela neurofibromatose de type 1 ou maladie de Recklinghausenest une pathologie autosomique dominante, par atteinte d’ungène suppresseur de tumeur situé sur le chromosome 17. Ilsurvient dans cette pathologie des neurofibromes multiples,des tumeurs primitives du système nerveux central (ménin-giomes, gliome du nerf optique, astrocytome, glioblastome,épendymome). Il peut survenir un phéochromocytome, desrhabdomyosarcomes. La présence de nodules de Lisch surl’iris est pathognomonique.La neurofibromatose de type 2 est également une pathologie
autosomique dominante, par atteinte d’un gène suppresseur detumeur situé sur le chromosome 22. Il existe des neurofibromesmultiples, en particulier au niveau du VIII, des méningiomes.Toute tumeur cérébrale peut entraîner par compression des voiesoptiques ou par hypertension intracrânienne une neuropathieoptique uni- ou bilatérale, pouvant se traduire par un œdèmepapillaire (Fig. 35), mais dans certains cas, l’examen clinique estnormal, et seule l’altération des PEV guide le diagnostic(Fig. 36). Il faut citer chez l’enfant le craniopharyngiome,tumeur bénigne, dérivée de l’épithélium pharyngé de la pochede Rathke (vestige du tractus pharyngohypophysaire primitif).Au stade évolué, elle peut entraîner une insuffisance hypophy-saire, des troubles visuels, un diabète insipide par atteintehypothalamique, une hypertension intracrânienne. Le traite-ment est chirurgical.• L’hypertension intracrânienne dite bénigne médicamenteuse
peut être secondaire, chez l’enfant, à la vitamine A, auxrétinoïdes, aux cyclines, à la ciclosporine, aux sulfamides.
Parmi les neuropathies optiques héréditaires, il convient dedistinguer les neuropathies isolées des neuropathies associéesà d’autres troubles. La neuropathie optique congénitalerécessive débute entre la naissance et 4 ans. Sa génétique estinconnue. La neuropathie optique liée au sexe récessive estégalement de génétique inconnue ; elle débute égalementdans la petite enfance. La neuropathie optique dominante deKjer débute entre 4 et 6 ans, et les anomalies génétiques sontsituées sur les chromosomes 3q et 18q. La neuropathieoptique de Leber est la mieux connue. Elle débute plutôtentre 15 et 35 ans, est de transmission maternelle mitochon-driale, avec trois types de mutations primaires, responsablesde 80 à 90 % de cette maladie : G3460A, T1484C etG11778A, cette dernière mutation étant de moins bonpronostic, avec des acuités visuelles très inférieures à 1/20,alors que les autres formes peuvent avoir une stabilisation à1 à 2/10. Faisons ici une parenthèse pour rappeler qu’unevision à 1 ou 2/10 peut éventuellement, avec aide, permettrede suivre une scolarité quasiment normale ; que la visionlégale pour conduire est de 6/10. Quelle que soit la patholo-gie oculaire, le discours initial est important. Il y a des degréstrès divers de malvoyance, des intégrations scolaires réussies,pour peu qu’on n’étiquette pas trop vite l’enfant malvoyantcomme handicapé profond, et que le personnel scolaire fasseun tant soit peu un effort pour accepter quelqu’un dedifférent. Revenons à la neuropathie optique de Leber ; il fautrechercher de façon systématique un bloc auriculoventricu-laire. Aucun traitement n’a fait la preuve de son efficacité,même si des traitements antioxydants sont parfois essayés.
• La liste des neuropathies héréditaires associées à d’autrespathologies neurologiques ou systémiques est longue. Citonsles maladies métaboliques comme les mucopolysaccharidoses,les lipidoses, les leucodystrophies ou le syndrome dePelizaeus-Merzbacher. Citons les neuropathies optiquesautosomiques dominantes souvent associées à divers degrésde surdité, survenant dans la première décennie de la vie, lesyndrome de Wolfram, associant à la neuropathie optique undiabète (insipide ou de type I). Citons les ataxies héréditaires(ataxie de Friedreich, ataxie spinocérébelleuse), les polyneu-ropathies héréditaires (syndrome de Charcot-Marie-Tooth,syndrome de Riley-Day), de nombreuses maladies mitochon-driales de l’enfant (syndrome mitochondrial encephalopathylactic acidosis stroke : MELAS, syndrome myoclonic epilepsyragged red fibers : MERFF, de Leigh, chronic progressive externalophthalmoplegia : CPEO, Kearns-Sayre syndrome : KSS).
Pathologie pupillaireL’anisocorie fait peur, car l’atteinte du III évoque toujours des
pathologies graves. Il faut, devant une anisocorie, raisonner de
Figure 35. Œdème papillaire témoin d’une hypertensionintracrânienne.
5 µV
Lobedroit
5 µV
Lobegauche
5 µV
Lobedroit
5 µV
Lobegauche
P1
N1
N2
P1
N1N2
N1
P1
P1
N1N2
N2
200 ms 200 ms
Lobe droitn°N1 84,2 4,3 86
144 11,9 86193 -9,8 73193 -9,8 73
P1N2N2
ms µV %
Lobe gauchen°N1 78,2 -13,0 86
147 14,5 86238 -17,1 82238 -17,1 82
P1N2N2
ms µV %
Lobe droitn°N1 126 -3,0 34
165 2,5 34186 0,0 37186 0,0 37
P1N2N2
ms µV %
Lobe gauchen°N1 132 -0,4 70
217 6,4 70253 -1,7 28253 -1,7 28
P1N2N2
ms µV %
Figure 36. Altération bilatérale des potentiels évoqués visuels, réalisés pour une baisse de vision progressive inexpliquée chez une enfant de 4 ans, ayantentraîné la réalisation d’une imagerie par résonance magnétique révélant un volumineux craniopharyngiome.
144414451446144714481449145014511452145314541455145614571458145914601461146214631464146514661467146814691470147114721473
147414751476147714781479148014811482148314841485148614871488148914901491149214931494149514961497149814991500150115021503150415051506150715081509151015111512151315141515151615171518
1519
15201521
.
4-120-A-10 ¶ Ophtalmologie infantile
18 Pédiatrie

façon posée et logique. Tout d’abord éliminer une pathologiepupillaire propre par l’examen ophtalmologique (uvéite, rupturesphinctérienne post-traumatique...). L’anisocorie physiologiqueest inférieure à 1 mm, elle est stable dans le temps, et quellesque soient les conditions d’éclairement. En cas d’anisocorieneurologique vraie, il faut déterminer quelle est la pupillepathologique.
Deux situations se présentent. Si l’anisocorie augmente à lalumière, il s’agit d’une atteinte du sphincter, et la pupillepathologique est la plus grande. Il s’agit d’une mydriaseparasympathique, puisque le sphincter irien est commandé parle III intrinsèque. Il s’agit le plus souvent chez l’enfant d’uneatteinte du premier neurone, s’associant volontiers à d’autressignes (III extrinsèque, céphalées...) et justifiant d’une imagerieen urgence, à la recherche de signes de méningite (les fibrespupillaires sont situées en position superficielle dans l’espacesous-arachnoïdien) ou de rupture d’anévrisme carotidien.L’atteinte du deuxième neurone ou pupille tonique d’Adie nesurvient qu’exceptionnellement chez l’enfant. Dans la deuxièmesituation, si l’anisocorie augmente à l’obscurité, il s’agit d’uneatteinte du dilatateur et la pupille pathologique est celle qui estla plus petite. Il s’agit d’un myosis sympathique, puisque ledilatateur de l’iris est commandé par le sympathique. Il fautrechercher les autres signes du syndrome de Claude-BernardHorner, et rechercher une compression sur la voie cervicalesympathique, par une radiographie cervicale (au mieux unscanner) englobant les apex pulmonaires ; chez l’enfant, il fautévoquer le neuroblastome.
Diplopie aiguëL’enfant va rarement se plaindre de diplopie. Cependant,
l’apparition brutale d’une diplopie binoculaire aiguë ou d’untrouble oculomoteur aigu, après avoir éliminé un strabismeaccommodatif en cas de strabisme convergent chez l’hypermé-trope (cf. supra), doit faire pratiquer une imagerie en urgencepour explorer le tronc cérébral.
■ Pathologie orbitopalpébrale
Malformations orbitairesNous reprenons la classification de Morax qui divise les
malformations craniofaciales en trois catégories [70].Les cranio-facio-sténoses sont consécutives à des sutures
prématurées des os du crâne et de la face. On distingue dans cecadre les plagiocéphalies, le syndrome d’Apert et le syndrome deCrouzon. Les complications ophtalmologiques sont liées àl’atteinte du nerf optique propre ou du fait de l’hypertensionintracrânienne possible, aux kératites d’exposition, au strabismeinduit avec éventuelle amblyopie consécutive. Les indicationschirurgicales, outre l’aspect esthétique, peuvent dépendre de lasurvenue de ces complications précitées.
La compréhension des fentes faciales doit beaucoup à PaulTessier. Elles entraînent des déficits osseux et tissulaires, maisaussi parfois des formations en excès. L’atteinte ophtalmologi-que est présente dans les fentes n° 3, 4 et 5, n° 6 et 8, qui,associées à une fente n° 7, peuvent constituer le syndrome deFranceschetti, et n° 9, 10 et 11.
L’hypertélorisme correspond au syndrome de Greig.
Malformations palpébrales et ptosiscongénital [71]
La pathologie malformative palpébrale est un vaste ensemblehétérogène par les présentations cliniques diverses, avec desmalformations mineures comme le colobome palpébral oul’ankyloblépharon, jusqu’aux formes plus sévères que consti-tuent l’ablépharie ou la cryptophtalmie.
Il peu exister des malpositions des paupières regroupantl’entropion et l’ectropion, ou bien des canthi, regroupantl’épicanthus ou le télécanthus. Le ptosis congénital isolé(Fig. 37, 38)
doit être distingué des blépharophimosis (Fig. 39), des ptosisneurogènes et des syndromes de rétraction. Toutes ces anoma-lies palpébrales peuvent être isolées ou rentrer dans la cadre desyndromes plus complexes avec origine génétique héréditaire.Les indications chirurgicales dépendent du risque fonctionnel(exposition cornéenne, amblyopie) et de l’esthétique.
Voies lacrymales [72]
En dehors de la pathologie malformative sévère dans le cadrede malformations craniofaciales plus sévères, la pathologiedominante chez le petit enfant est l’imperforation des voieslacrymales simple, sans anomalie anatomique décelable, corres-pondant à une sténose ou un rétrécissement localisé soit auxvoies horizontales (canalicules lacrymaux et canal d’union), soità la voie verticale (sténose le plus souvent du canal lacrymona-sal au niveau de la valvule de Hasner qui relie les voieslacrymales à la cavité nasale).
La symptomatologie d’une imperforation simple des voieslacrymales chez le nourrisson se traduit par un larmoiementclair bilatéral avec épisodes itératifs de surinfections à type deconjonctivites purulentes.• Avant 4 mois, chaque épisode infectieux est traité de façon
ponctuelle par collyre antibiotique.
Figure 37. Ptosis préopératoire.
Figure 38. Ptosis postopératoire.
Figure 39. Blépharophimosis.
1522152315241525152615271528152915301531153215331534153515361537153815391540154115421543154415451546154715481549
1550
155115521553155415551556
1557
1558
155915601561156215631564156515661567156815691570157115721573157415751576
1577
1578
157915801581158215831584158515861587
158815891590159115921593
1594
159515961597159815991600160116021603160416051606160716081609
.
.
Ophtalmologie infantile ¶ 4-120-A-10
19Pédiatrie

• Entre 4 mois et 10 à 12 mois, on peut proposer un sondagedes voies lacrymales, chez un enfant vigile, avec contention.
• En cas d’échec de la procédure, il faut proposer une anesthé-sie générale avec intubation provisoire des voies lacrymalespendant 2 à 3 mois par une sonde en silicone, qui est retiréesimplement en consultation.
• Après 10 à 12 mois, l’anesthésie est nécessaire chez le jeuneenfant qui se débat... On réalise donc une intubation provi-soire quasiment systématiquement.En cas d’échec de la procédure, on peut proposer une
dacryo-cysto-rhinostomie.La dacryocystite est une infection du sac lacrymal, se tradui-
sant par une tuméfaction au canthus interne, chaude (Fig. 40) ;il n’y a pas forcément de fièvre. Le traitement antibiotique estlocal et général, avec à distance sondage et intubation de la voielacrymale, voire dacryocystorhinostomie. Il faut différencierchez l’enfant de moins de 1 an la « simple » dacryocystite de laplus redoutable ostéite du malaire. Une ethmoïdite a, quant àelle, une symptomatologie rhinopharyngée associée, parfois uneexophtalmie inflammatoire, souvent une fièvre plus importante.
Conduite à tenir devant un chalazionet devant un orgelet
La chalazion est une pathologie très fréquente, se traduisantpar une tuméfaction arrondie de la paupière, pouvant prédomi-ner sur le bord cutané ou sur le bord conjonctival. Il s’agitd’une obstruction par un sébum épaissi d’une des glandes deMeibomius de la paupière (celles-ci sécrètent normalement unsébum fluide correspondant à la phase lipidique du filmlacrymal). La glande obstruée a tendance à gonfler et former unpetit kyste qui se surinfecte souvent. Le traitement repose surune pommade antibiocorticoïde locale à instiller dans le cul-de-sac palpébral inférieur pour une meilleure diffusion, pendantéventuellement plusieurs semaines, avec parfois nécessité detraitements itératifs en cas de récidive. Il faut y associer desmassages doux des paupières. En cas d’échec d’un traitementbien conduit et au besoin renouvelé, on peut proposer uneincision chirurgicale.
Un orgelet est une pathologie primitivement infectieuse de laracine d’un cil, souvent à staphylocoque, se traduisant par unedouleur palpébrale, avec à l’examen un aspect de kyste rouge àla racine d’un cil. Le traitement repose sur une antibiothérapielocale.
■ Pathologie tumorale
Rétinoblastome [23]
Il est peu de cas en ophtalmologie où le pronostic vital estengagé. Le rétinoblatome fait partie de ces cas, et le premier
signe d’un rétinoblastome n’est parfois pas la classique leucoco-rie (signant une tumeur déjà évoluée), mais une simple dévia-tion oculaire (l’œil atteint malvoyant ne prenant pas lafixation). Le rétinoblastome est une tumeur se développant auxdépens des cônes. Son incidence est de 1 pour 15 000 à 20 000naissances. Il prend au fond d’œil un aspect blanchâtre surélevé,avec une ou plusieurs localisations (Fig. 41).
Il peut être uni- ou bilatéral. La classification en cinq stadesest celle de Reese-Ellsworth. La présence de calcifications auscanner est quasiment pathognomonique (Fig. 42) (le seuldiagnostic différentiel pouvant être évoqué étant une infectionà Toxocara canis, survenant dans un contexte différent ; lessérologies peuvent aider au diagnostic). Le modèle d’uneatteinte d’un gène suppresseur de tumeur évoqué par Knudsonen 1971 a été confirmé par les progrès ultérieurs de la généti-que. Le gène en cause dans la physiopathogénie du rétinoblas-tome est RB1, situé en 13q14, gène codant pour une protéineintervenant dans le cycle cellulaire et bloquant le passage enphase S. Le défaut du gène RB1 peut correspondre à unemutation, dont de très nombreuses sont identifiées, ou à uneabsence par microdélétion par exemple. Dans les cas familiauxde rétinoblastome (10 à 15 % des cas), une mutation constitu-tionnelle est transmise selon un mode autosomique dominant,avec développement tumoral en cas d’événement à type denouvelle mutation sur le second allèle du gène RB1 ; le risquede tumeurs multiples, de pinéaloblastome, mais aussi d’autrestumeurs comme les sarcomes ou les mélanomes, est grand. Lescas sporadiques regroupent d’une part les cas de mutationconstitutionnelle apparue dans l’un des gamètes des deuxparents, avec l’ensemble des cellules de l’organisme atteintes,donc un risque majeur de tumeurs multiples, et d’autre part lescas dans lesquels deux mutations sont survenues de façonfortuite au niveau somatique sur une cellule, avec développe-ment d’une tumeur unilatérale unique (rappelons cependantque le risque de présenter une mutation constitutionnelle,même en cas de tumeur unilatérale, est estimé à 10 %).
Le traitement du rétinoblastome repose sur la chirurgie desformes évoluées (stades 4 et 5 de Reese), c’est-à-dire l’énucléa-tion, voire l’exentération. La chirurgie mutilante n’est en règleproposée, sauf exception, que dans les formes unilatéralesévoluées. Les différentes autres armes thérapeutiques sont lachimiothérapie, la radiothérapie, la photocoagulation, lathermothérapie. Le traitement conservateur doit être proposé le
Figure 40. Dacryocystite droite.
Figure 41. Rétinoblastome : aspect du fond d’œil.
Figure 42. Rétinoblastome bilatéral avec calcifications orbitaires.
16101611161216131614161516161617161816191620162116221623162416251626162716281629
1630
1631
16321633163416351636163716381639164016411642164316441645164616471648164916501651
1652
1653
16541655
1656165716581659166016611662
16631664166516661667166816691670167116721673167416751676167716781679168016811682168316841685168616871688168916901691
1692169316941695169616971698
.
4-120-A-10 ¶ Ophtalmologie infantile
20 Pédiatrie

plus souvent possible. Il faut distinguer, dans la décisionthérapeutique, le pronostic vital du pronostic fonctionnel.
La surveillance du sujet atteint et de sa fratrie est bienentendu fondamentale ; différents calculs de probabilité deprédisposition au rétinoblastome ont été établis et permettentde proposer une fréquence adaptée de surveillance. Le conseilgénétique et l’étude chez le sujet atteint et dans sa famille dugène RB1, soit par étude moléculaire directe, soit par étudeindirecte, ou encore par étude cytogénétique (en premièreintention en cas de syndrome polymalformatif), peut permettredans certains cas d’exclure certains sujets de la fratrie d’unenfant atteint d’un risque accru de rétinoblastome.
Tumeurs orbitairesElles sont multiples, de présentations cliniques variées, de
circonstances de découverte diverses : masse directement visible,exophtalmie, diplopie ou strabisme, baisse d’acuité visuelle,anisocorie, examen du fond d’œil faisant apparaître uneanomalie papillaire (pâleur, œdème papillaire). L’imagerie estbien entendu très contributive du diagnostic. Le contexteclinique oriente bien entendu. Nous rappellerons qu’à ladifférence de l’adulte, les tumeurs primitives de l’orbite sontrares et qu’il faut évoquer plus souvent des localisationssecondaires (leucémies, neuroblastome). Parmi les tumeursprimitives, nous citerons le rhabdomyosarcome, le gliome dunerf optique pouvant entrer dans le cadre d’une neurofibroma-tose, les lymphangiomes (Fig. 43), les hémangiomes, les lym-phomes, les maladies histiocytaires... Citons ici des tumeurscongénitales bénignes : le kyste épidermoïde de la queue dusourcil (Fig. 44) et le kyste épidermoïde du limbe (Fig. 45).
Hémangiomes de la faceIl faut distinguer les hémangiomes plans (tel celui présent
dans le syndrome de Sturge-Weber-Krabbe) des hémangiomestubéreux, en particulier pour l’ophtalmologiste les hémangio-mes palpébraux dont le risque est l’amblyopie par privation
visuelle unilatérale (Fig. 46). Les volumineux hémangiomespeuvent faire évoquer un syndrome PHACES, associant desmalformations de la fosse postérieure, des hémangiomes, desanomalies artérielles, une coarctation de l’aorte, des anomaliesoculaires à type d’hémangiome choroïdien, de cataracte... (eyes),de malformations sternales (Fig. 47).
■ TraumatologieConduite à tenir devant une contusionoculaire
En cas de contusion oculaire, il faut rechercher des signes degravité : épistaxis, emphysème sous-cutané, troubles oculomo-teurs. Il ne faut pas hésiter à demander un scanner (sansurgence immédiate), bien plus performant que les radiographiesle plus souvent inutiles. Il faut se méfier des troubles d’appari-tion secondaire. Un fond d’œil est à réaliser dans les 48 heures ;des photopsies persistantes au même endroit font évoquer unedéchirure rétinienne à photocoaguler au laser ; une baissed’acuité brutale évoque, soit un décollement de rétine, soit unœdème maculaire de Berlin.
Figure 43. Lymphangiome orbitaire droit responsable d’un strabismeet d’une amblyopie gauches.
Figure 44. Kyste épidermoïde de la queue du sourcil, vueperopératoire.
Figure 45. Kyste dermoïde du limbe bilatéral.
Figure 46. Hémangiome palpébral gauche obstructif responsabled’une amblyopie de déprivation.
Figure 47. Hémangiome fronto-temporo-palpébral droit. Suspicion desyndrome PHACES : malformations de la fosse postérieure, hémangio-mes, anomalies artérielles, coarctation de l’aorte, anomalies oculaires àtype d’hémangiome choroïdien, de cataracte... (eyes), malformationssternales.
169917001701170217031704170517061707170817091710
1711
1712171317141715171617171718171917201721172217231724172517261727
1728
1729173017311732
173317341735173617371738
1739
1740
1741
1742174317441745174617471748174917501751
Ophtalmologie infantile ¶ 4-120-A-10
21Pédiatrie

Fracture du plancher de l’orbiteElle survient après une contusion oculaire. La diplopie en
position primaire est inconstante. Le signe capital est lalimitation de l’élévation par incarcération du muscle droitinférieur. Le scanner est à réaliser en urgence, et l’interventionchirurgicale est à réaliser dans les 48 heures du fait du risque defibrose, soit par voie conjonctivale, soit par voie cutanée(Fig. 48–50).
Plaies transfixiantes du globeLes plaies transfixiantes sont à suspecter selon les circonstan-
ces du traumatisme. À l’examen la cornée et/ou la conjonctiveprennent la fluorescéine. Il peut exister un phénomène deSeidel (dilution de la fluorescéine par l’humeur aqueuse sortantde la chambre antérieure). La chambre antérieure est inflamma-toire, il peut exister un hyphéma, une cataracte traumatique(Fig. 51). L’enfant est à adresser rapidement en milieu spécialisépour un examen au microscope plus ou moins une explorationsous anesthésie générale.
Plaie de paupièreUne plaie de paupière est à suturer en urgence si elle est
superficielle, par exemple à la soie 6.0. Il faut se méfier d’uneplaie des voies lacrymales si la plaie se situe au canthus interne,ce qui alors nécessite une exploration des voies lacrymales avecéventuelle intubation par une sonde en silicone (Fig. 52). Il fautse méfier d’une plaie du releveur de la paupière supérieure si laplaie est profonde ; ce muscle est à tester simplement en faisant
regarder le patient vers le haut. Une plaie du bord libre est àsuturer rapidement, sous microscope en milieu spécialisé, avecaffrontement précis des bords, pour le résultat esthétique etfonctionnel ultérieur.
Brûlures oculairesL’exposition des yeux non protégés à la neige, du fait de la
réverbération majeure, entraîne une kératite diffuse superficielle,très douloureuse bien que rarement sévère ; le traitement reposesur un cicatrisant local et la protection des UV. L’expositionsolaire propre, quant à elle (fixation du soleil, par exemplependant les phénomènes d’éclipse), peut engendrer une brûluremaculaire avec destruction localisée de photorécepteurs pouvantrésulter en un scotome central.
La projection d’un liquide ou gel (souvent ménager) dans lesyeux est d’une gravité qui dépend du pH du produit. Lesproduits acides entraînent une brûlure superficielle, du faitd’une coagulation des protéines de surface ; cela dit, la brûlurepeut être grave ; le traitement repose sur les lavages abondants,une antibiothérapie locale et des pommades cicatrisantes. Lesbrûlures basiques sont, quant à elles, potentiellement gravissi-mes, avec risque de perforation oculaire ; l’antibiothérapie localeet souvent systémique est requise, associée aux lavages itératifsabondants et au traitement lubrifiant local. Une prise en chargespécialisée avec surveillance rapprochée à la recherche d’unamincissement cornéen est indispensable.
Figure 48. Fracture du plancher de l’orbite gauche ; limitation del’élévation.
Figure 49. Fracture du plancher de l’orbite gauche. Scanner.
Figure 50. Fracture du plancher de l’orbite gauche. j5 postopératoire.
Figure 51. Plaie perforante de cornée.
Figure 52. Traumatisme par un pinceau. Se méfier : trajet de la voielacrymale.
1752
1753175417551756175717581759
1760
176117621763176417651766176717681769
1770
1771177217731774177517761777
1778177917801781
1782
17831784178517861787178817891790
179117921793179417951796179717981799180018011802
4-120-A-10 ¶ Ophtalmologie infantile
22 Pédiatrie

■ Uvéites
Uvéites infectieuses : herpès,cytomégalovirus, toxoplasmose,toxocarose...
L’uvée est le tissu oculaire correspondant en avant à l’iris, àla partie intermédiaire de l’œil au corps ciliaire, et au niveau dusegment postérieur à la choroïde. Ces trois éléments sont encontinuité. On regroupe sous le vocable « uvéite » un ensemblede pathologies inflammatoires et infectieuses.
Toute infection intraoculaire entraîne le plus souvent uneréaction inflammatoire se traduisant par une uvéite. Celle-cipeut être antérieure, avec réaction inflammatoire de chambreantérieure se traduisant par un effet Tyndall (c’est-à-direprésence de protéines ou de cellules dans la chambre antérieurede l’œil), par des précipités rétrodescemétiques (Fig. 53), par dessynéchies entre l’iris et le cristallin, par une hypertonie oculaire.L’uvéite intermédiaire se traduit par un infiltrat inflammatoirede la pars plana. L’uvéite postérieure se traduit par une réactioninflammatoire du corps vitré qui occupe tout le segmentpostérieur de l’œil : il s’agit d’une hyalite. L’atteinte rétinienneou choroïdienne se traduit au fond d’œil par une lésion le plussouvent visible, parfois diagnostiquée par l’imagerie : angiogra-phie à la fluorescéine ou au vert d’indocyanine. L’atteinteinflammatoire du nerf optique se traduit par une papillite(Fig. 54).
Les étiologies d’uvéite infectieuse sont très nombreuses, et setraduisent par des caractères cliniques plus ou moins évocateurs.
Une atteinte herpétique se traduit par une uvéite antérieureavec endothélite se manifestant par des plis de la membrane deDescemet ; il existe volontiers une hypertonie oculaire, deszones d’atrophie irienne en cas de caractère récidivant. Il fautrechercher au fond d’œil un foyer de rétinite active.
Le cytomégalovirus entraîne une rétinite, chez le nouveau-néen cas d’infection maternofœtale, ou bien chez l’enfant plusgrand dans les cas d’immunodépression.
La toxoplasmose est fréquente ; elle peut être congénitale.Dans la majorité des cas (80 % des sujets ont une sérologiepositive), elle se traduit chez l’enfant comme chez l’adulte par
des épisodes récurrents de rétinite, avec foyer blanchâtre actif,souvent en contiguïté avec un foyer pigmenté ancien (Fig. 55).L’indication d’un traitement antiparasitaire dépend de lalocalisation du foyer : un foyer central menaçant l’acuitévisuelle doit être traité, alors que le traitement d’un foyerpériphérique peut se discuter.
Toxocara canis se traduit par une uvéite postérieure chez unenfant de 3 à 10 ans, souvent en contact dans son environne-ment domestique avec un chien. L’aspect blanchâtre pseudotu-moral avec tractions vitréennes, et surtout parfois calcifications,peut en imposer pour un rétinoblastome. L’âge, le contexte,l’hyperéosinophilie, les sérologies, redressent le diagnostic. Letraitement peut nécessiter une chirurgie à type de vitrectomie.
Arthrite juvénile idiopathiqueLes uvéites associées à des arthralgies intéressent d’autant plus
l’ophtalmologiste qu’elles n’intéressent pas le pédiatre, etinversement les arthralgies associées à des uvéites intéressentd’autant plus le pédiatre qu’elles n’intéressent pas l’ophtalmo-logiste. En effet, au sein du spectre des pathologies rhumatolo-giques associées à des uvéites, l’atteinte ophtalmologique estd’autant plus sévère que l’atteinte rhumatologique est minime,et inversement. Le lecteur pardonnera ce raccourci, mais ilreflète en fait la difficulté qu’ont souvent les équipes gérant lesarthrites juvéniles idiopathiques, dans lesquelles l’atteinteophtalmologique prédomine : l’ophtalmologiste « dicte » parfoisl’indication d’une augmentation de la corticothérapie systémi-que, voire demande l’introduction d’un immunosuppresseurpour une aggravation de l’état oculaire, sans savoir forcémentgérer ces thérapeutiques, d’où son besoin du pédiatre.
Revenons à une classification plus didactique. On distingue laforme systémique de la maladie (maladie de Still), dans laquellel’atteinte ophtalmologique est exceptionnelle. La forme polyar-ticulaire est également une forme dans laquelle l’atteinteophtalmologique est rare. Au contraire, la forme oligo- oumonoarticulaire, touchant plutôt la petite fille, entraîne uneuvéite dans environ la moitié des cas. Il s’agit d’une uvéite nongranulomateuse, souvent torpide, pouvant précéder l’atteintearticulaire. Elle se complique de kératite en bandelettes, desynéchies iridocristalliniennes, de cataracte, d’œdème maculairecystoïde. Le pronostic fonctionnel est donc menacé. Un traite-ment par corticothérapie systémique, par méthotrexate, ou pard’autres types d’immunosuppresseurs peut être nécessaire,adjoint au traitement local corticoïde et cycloplégique. Il s’agitde ces cas ou l’ophtalmologiste a recours au pédiatre.
Autres étiologies d’uvéiteIl s’agit de pathologies inflammatoires comme la sarcoïdose,
les spondylarthropathies, la maladie de Vogt-Koyanagi-Harada...Il peut s’agir de nombreuses étiologies infectieuses : syphilis,tuberculose, maladie de Lyme...
■ ConclusionAu terme de ce panorama, nous mesurons avec une acuité
encore plus aiguë la difficulté de la tâche consistant à évoquer
Figure 53. Uvéite antérieure granulomateuse.
Figure 54. Papillite droite. Aspect angiographique.
Figure 55. Toxoplasmose oculaire.
1803
1804
1805
1806
1807180818091810181118121813181418151816181718181819182018211822182318241825182618271828182918301831183218331834183518361837183818391840
1841184218431844184518461847184818491850185118521853
1854
185518561857185818591860186118621863186418651866186718681869187018711872187318741875187618771878187918801881188218831884
1885
1886188718881889
1890
18911892
Ophtalmologie infantile ¶ 4-120-A-10
23Pédiatrie

en quelques lignes l’ensemble de l’ophtalmologie infantile. Ilexiste de nombreux manques, certainement. Le lecteur nousaccordera son indulgence.
Nous ne pouvons qu’insister sur le caractère passionnant del’ophtalmologie, et bien entendu de l’ophtalmologie infantile.La prise en charge d’un enfant en ophtalmologie est spécifique.Rappelons de nouveau que la fonction visuelle est en dévelop-pement après la naissance. Rappelons que toute pathologieprécoce, que tout doute ou suspicion sur une anomalie ophtal-mologique doit requérir un examen avec un fond d’œil. Nousterminerons sur l’idée, dans les cas où malheureusement noshumbles armes médicales ne permettent pas d’éviter à unenfant la malvoyance, en cas de pathologie sévère bilatérale, surla nécessité d’une prise en charge spécifique (dans de nombreu-ses structures adaptées) afin que son éveil sensoriel, même grevépar une mauvaise vision, puisse être assuré au mieux.
■ Références[1] Allikmets R, Shroyer NF, Singh N, Seddon JM, Lewis RA,
Bernstein PS, et al. Mutation of the Stargartdt disease gene (ABCR) inage-related macular degeneration. Science 1997;277:1805-7.
[2] Bailly A. Dictionnaire Grec-Français. Paris: Hachette; 1963.[3] Dictionnaire « Garnier-Delamare » des termes de médecine. Paris:
Maloine; 1992.[4] Worth CA. Squint: Its cause, pathology and treatment. Philadelphia:
Blakiston; 1903.[5] Orssaud C, Dufier JL.Amblyopie. EMC (Elsevier Masson SAS, Paris),
Ophtalmologie, 21-595-A-10, 1998.[6] Bui Quoc E. Fondements de la notion de période sensible du dévelop-
pement visuel. EMC (Elsevier Masson SAS, Paris), Ophtalmologie,21-592-A-05, 2006.
[7] Speeg-Schatz C. Développement des fonctions visuelles chez le jeuneenfant. Ann Pediatr (Paris) 1996;43:372-8.
[8] Tychsen L. Binocular vision. In: Adler’s physiology of the eye. St Louis:CV Mosby; 1992. p. 773-853.
[9] Romano PE, Romano JA, Puklin JE. Stereoacuity development inchildren with normal binocular single vision. Am J Ophthalmol 1975;79:966-71.
[10] Atkinson J, Braddick O. Stereoscopy discrimination in infants. Percep-tion 1976;5:29-38.
[11] Atkinson J, Braddick O, Moar K. Development of contrast sensitivityover the first 3 months of life in the human infant. Vision Res 1977;17:1037-44.
[12] Banks MS. The development of spatial and temporal contrastsensitivity. Curr Eye Res 1982;2:191-8.
[13] Teller DY. Visual development. Vision Res 1986;26:1483-506.[14] Committee on Practice and Ambulatory Medicine Section on
Ophthalmology American. Eye examination in infants, children, andyoung adults by pediatricians: organizational principles to guide anddefine the child health care system and/or improve the health of allchildren. Association of Certified Orthoptists; American Associationfor Pediatric Ophthalmology and Strabismus; American Academy ofOphthalmology. Ophthalmology 2003;110:860-5.
[15] Committee on Practice and Ambulatory Medicine. Section onOphthalmology. American Association of Certified Orthoptists;American Association for Pediatric Ophthalmology and Strabismus;American Academy of Ophthalmology. Eye examination in infants,children, and young adults by pediatricians. Pediatrics 2003;111(4Pt1):902-7.
[16] Weinstock VM, Weinstock DJ, Kraft SP. Screening for childhoodstrabismus by primary care physicians. Can Fam Physician 1998;44:337-43.
[17] Bourcier-Bareil F, Lecuyer AI, Burel B, Delplace MP. Intérêt du Bébé-Vision dans le dépistage de masse de l’amblyopie strabique etanisométropique du nourrisson. J Fr Ophtalmol 2001;24:1034-9.
[18] Denis D, Benso C, Wary P, Fogliarini C. La réfraction chez l’enfant :épidémiologie, évolution, évaluation et mode de correction desamétropies. J Fr Ophtalmol 2004;27:943-52.
[19] Adenis JP, Salomon JL, Saint-Blancat P. Conjonctivites bactériennes.EMC (Elsevier Masson SAS, Paris), Ophtalmologie, 21-130-B-20,1994 : 7p.
[20] Fardeau C. Conjonctivites infectieuses virales. EMC (Elsevier MassonSAS, Paris), Ophtalmologie, 21-130-D-10, 1995 : 5p.
[21] Bloch-Michel E, Helleboid L. Conjonctivites allergiques. EMC(Elsevier Masson SAS, Paris), Ophtalmologie, 21-130-E-10, 1994 :10p.
[22] Dighiero P, Ellies P, Grateau G, D’Hermies F, PouliquenY, Legeais JM,et al. Mise au point sur les dystrophies de cornée. J Fr Ophtalmol 1999;22:226-33.
[23] Dufier JL, Kaplan J. Œil et génétique. Rapport de la Société Françaised’Ophtalmologie. Paris: Masson; 2005.
[24] Roche O, Beby F, Orssaud C, Dupond Monod S, Dufier JL. Cataractecongénitale. J Fr Ophtalmol 2006;29:443-55.
[25] Foster A, Gilbert C. Cataract in children. Acta Paediatr 2003;92:1376-8.
[26] Francis PJ, Berry V, Bhattacharya SS, Moore AT. The genetics ofchildhood cataract. J Med Genet 2000;37:481-8.
[27] Faivre L, Megarbane A, Alswaid A, Zylberberg L, Aldohayan N,Campos-Xavier B, et al. Homosigosity mapping of a Weill-Marchesanisyndrome locus to chromosome 19p13.3-p13.2. Am J Med Genet A2003;123:204-7.
[28] Bui Quoc E, Roche O, Hakiki S, Dufier JL. Prise en charge de larétinopathie du prématuré en service spécialisé. J Fr Ophtalmol 2004;27:883-9.
[29] Ashton, Ward B, Serpell G. Effect of oxygen on developing retinalvessels with particular reference to the problem of retrolentalfibroplasias. Br J Ophthalmol 1954;38:397-430.
[30] Ryzman P, Hamard H, Mondon H, Lefrancois A, Relier JP,Minkowski A, et al. Données actuelles sur la rétinopathie aiguë du pré-maturé sous oxygénothérapie. J Fr Ophtalmol 1981;4:565-70.
[31] Gerhard JP. La prévention de la rétinopathie du prématuré. J FrOphtalmol 1986;9:583-96.
[32] Larsson E, Holmstrom G. Screening for retinopathy of prematurity:evaluation and modification of guidelines. Br J Ophthalmol 2002;86:1399-402.
[33] Andruscavage L, Weissgold DJ. Screening for retinopathy ofprematurity. Br J Ophthalmol 2002;86:1127-30.
[34] Thouvenin D, Legavre L, Bourdiol AM, Arne JL. Rétinopathie de laprématurité. Intérêt actuel de la surveillance des prématurés et nouveau-nés à risques. J Fr Ophtalmol 1992;15:191-7.
[35] Miyamoto N, Mandai M, Takagi H, Suzuma I, Suzuma K, Koyama S,et al. Contrasting effect of estrogen on VEGF induction under differentoxygen status and its role in murine ROP. Invest Ophthalmol Vis Sci2002;43:2007-14.
[36] The Committee for the Classification of Retinopathy of Prematurity.An international classification of retinopathy of prematurity. ArchOphthalmol 1984;102:1130-4.
[37] The International Committee for the Classification of the Late Stages ofRetinopathy of Prematurity. An international classification ofretinopathy of prematurity. II. The classification of retinal detachment.Arch Ophthalmol 1987;105:906-12.
[38] The International Committee for the Classification of the Late Stages ofRetinopathy of Prematurity. An international classification ofretinopathy of prematurity. II. The classification of retinal detachment.Erratum. Arch Ophthalmol 1987;105:1498.
[39] Dollfus H, Ghazi I, Dufier JL. Rétinopathies pigmentaires. EMC(Elsevier Masson SAS, Paris), Ophtalmologie, 21-243-D-10, 1997 :12p.
[40] Bonnet M. Le syndrome de Coats. J Fr Ophtalmol 1980;3:57-66.[41] Rosberger DF, Goldberg M. Hereditary vitreoretinopathies. In:
Ttrabousli E, editor. Genetic diseases of the eye. Oxford: OxfordUniversity Press; 1998. p. 435-60.
[42] Speeg-Schaatz C, Le Marec B. Erreurs génétiques et œil. In: Œil etpathologie générale. Rapport de la SFO. Paris: Masson; 1997. p. 3-80.
[43] Zech JC, Trepsat C. Dégénérescences vitréo-rétiniennes héréditaires.EMC (Elsevier Masson SAS, Paris), Ophtalmologie, 21-248-A-20,1997 : 10p.
[44] Wagner H. Ein bisher unbekanntes Erbleiden des Auges (Degeneratiohyaloido-retinalis hereditaria), beobachtet im Kanton Zurich. KlinMonatsbl Augenheilkd 1938;100:840-57.
[45] Waxman SL, Bergen RL. Wagner’s vitreoretinal degeneration. AnnOphthalmol 1982;12:1150-5.
[46] Gupta SK, Leonard BC, Damji KF, Bulman DE.Aframe shift mutationin a tissue specific alternatively spliced exon of collagen 2AinWagner’svitreoretinal degeneration. Am J Ophthalmol 2002;133:203-10.
1893
1894
1895
1896
1897
1898
1899
1900
1901
1902
1903
1904
1905
1906
1907
1908
1909
19101911191219131914191519161917191819191920192119221923192419251926192719281929193019311932193319341935193619371938193919401941194219431944194519461947194819491950195119521953195419551956195719581959196019611962
19631964196519661967196819691970197119721973197419751976197719781979198019811982198319841985198619871988198919901991199219931994199519961997199819992000200120022003200420052006200720082009201020112012201320142015201620172018201920202021202220232024202520262027202820292030203120322033203420352036
.
4-120-A-10 ¶ Ophtalmologie infantile
24 Pédiatrie

[47] Stickler GB, Belau PG, Farrell FJ, Jones JD, Pugh DG, Steinberg AG,et al. Hereditary progressive arthro-ophthalmopathy. Mayo Clin Proc1965;40:433-55.
[48] François J, De Rouck A, Cambie E. Dégénérescence hyaloïdéo-tapéto-rétinienne de Goldmann et Favre. Ophthalmologica 1974;168:81-96.
[49] Criswick VG, Schepens CL. Familial exsudative vitreoretinopathy. AmJ Ophthalmol 1969;68:578-94.
[50] Spranger J, Langer LO, Wiedemann HR. Bone dysplasias.Stuttgart/Philadelphia: Fischer/Saunders; 1974.
[51] Marshall D. Ectodermal dysplasia: report of a kindred with ocularabnormalities and hearing defect. Am J Ophthalmol 1958;45:143-56.
[52] Taillard F, Desbois JC, Delepine N,Allaneau C, Wyart D. Syndrome deMarshall ou syndrome de Stickler? Discussion à propos d’une famille.Ann Pediatr (Paris) 1987;34:279-84.
[53] Chemke J, Carmi R, Galil A, Bar-Ziv Y, Ben-Ytzhak I, Zurkowski L.Weissenbacher-Zweymuller syndrome: a distinct autosomal recessiveskeletal dysplasia. Am J Med Genet 1992;43:989-95.
[54] Haas J. Über das zusammenvorkommen von Veränderungen der Retinaund Chorioidea. Arch Augenheilkd 1898;37:343-8.
[55] George ND, Yates JR, Moore AT. Clinical features in affected maleswith X-linked retinoschisis. Arch Ophthalmol 1996;114:274-80.
[56] Snead MP, Yates JRW. Clinical and molecular genetics of Stickler syn-drome. J Med Genet 1999;36:353-9.
[57] Vintiner GM, Temple IK, Middleton-Price HR, Baraitser M,Malcolm S. Genetic and clinical heterogeneity of Stickler syndrome.Am J Med Genet 1991;41:44-8.
[58] Ahmad NN, McDonald-McGinn DM, Dixon P, Zackai EH,Tasman WS. PCR assay confirms diagnosis in syndrome with variablyexpressed phenotype: mutation detection in Stickler syndrome. J MedGenet 1996;33:678-81.
[59] Wilkin DJ, Mortier GR, Johnson CR, Jones MC, De Paepe A, Shoat M,et al. Correlation of linkage data with phenotype in eight families withStickler syndrome. Am J Med Genet 1998;80:121-7.
[60] RichardsAJ, Baguley DM, Yates JR, Lane C, Nicol M, Harper PS, et al.Variation in the vitreous phenotype of Stickler syndrome can be causedby different amino acid substitutions in the X position of the type IIcollagen Gly-X-Y triple helix. Am J Hum Genet 2000;67:1083-94.
[61] RichardsAJ, Martin S,Yates JR, Scott JD, Baguley DM, Pope FM, et al.COL2A1 exon 2 mutations: relevance to the Stickler and Wagner syn-dromes. Br J Ophthalmol 2000;84:364-71.
[62] Fritsch D, Vallat M, Lagoutte F, Verin P. Prévention du décollement derétine dans le syndrome de Wagner-Stickler. Bull Soc Ophtalmol Fr1989;89:657-64.
[63] Monin C, Van Effenterre G, Andre-Sereys P, Haut J. Prophylaxie dudécollement de rétine de la maladie de Wagner-Stickler. J Fr Ophtalmol1994;17:167-74.
[64] Rigaudière F, Leid J, Viénot F, Le Gargasson JF. Comprendre et testerles déficiences de la vision des couleurs chez l’enfant. J Fr Ophtalmol2006;29:87-102.
[65] Bui Quoc E. Strabisme chez l’enfant. EMC (Elsevier Masson SAS,Paris), Pédiatrie, 4-120-D-10, 2004.
[66] Thouvenin D. Strabismes précoces. EMC (Elsevier Masson SAS,Paris), Ophtalmologie, 21-550-A-02, 2002 : 8p.
[67] Péchereau A. Strabisme de l’enfant. Rev Prat 2003;53:1827-33.[68] Klainguti G. Strabisme accommodatif. EMC (Elsevier Masson SAS,
Paris), Ophtalmologie, 21-550-A-03, 2001.[69] Spielmann A. Les strabismes. De l’analyse clinique à la synthèse chi-
rurgicale. Paris: Masson; 1989 (262p).[70] Morax S, Hamédani M. Malformations craniofaciales. EMC (Elsevier
Masson SAS, Paris), Ophtalmologie, 21-100-D-30, 2001 : 9p.[71] Utéza Y, Dufier JL. Affections génétiques et congénitales des paupiè-
res. EMC (Elsevier Masson SAS, Paris), Ophtalmologie, 21-100-A-10,2001 : 13p.
[72] Urvoy M, Toulemont PJ, Aubert V, Le Callonnec A. Pathologielacrymale chez le nourrisson et l’enfant. EMC (Elsevier Masson SAS,Paris), Ophtalmologie, 21-170-B-30, 2006.
E. Bui Quoc, Chef de clinique des Universités, assistant des hôpitaux de Paris ([email protected]).Service d’ophtalmologie du Professeur Jean-Louis Dufier, Hôpital Necker-Enfants-Malades, 149, rue de Sèvres, 75015 Paris, France.
Toute référence à cet article doit porter la mention : Quoc E. Bui Ophtalmologie infantile. EMC (Elsevier Masson SAS, Paris), Pédiatrie, 4-120-A-10, 2007.
Disponibles sur www.emc-consulte.com
Arbresdécisionnels
Iconographiessupplémentaires
Vidéos /Animations
Documentslégaux
Informationau patient
Informationssupplémentaires
Auto-évaluations
203720382039204020412042204320442045204620472048204920502051205220532054205520562057205820592060206120622063206420652066206720682069
210321042105
2106
207020712072207320742075207620772078207920802081208220832084208520862087208820892090209120922093209420952096209720982099210021012102
Ophtalmologie infantile ¶ 4-120-A-10
25Pédiatrie





























