Embed Size (px)
Citation preview

Geneviève Fortier
SYNTHÈSES ÉNANTIOSELECTIVES DE ~ÉTRAHYDROFURANES D'ORIGINE MARINE
Thèse présentée
à la Faculté des études supérieures pour l'obtention
du grade de Philosophiae Doctor (Ph.D.)
Département de chimie FACULTE DES SCIENCES ET DE GÉNIE
UNIVERSITÉ LAVAL QUÉBEC
MAI 1998
O Geneviève Fortier, 1998

National Library BbWhèque nationale du Canada
Acquisitions and Acquisitions et Bibliog raphic Services services bibliographiques
395 W e û i i o n Street 395, nie Wellington OtîawaON K 1 A W OttawaON K1AûN4 Canada canada
The author has granted a non- L'auteur a accordé une licence non exclusive licence allowing the exclusive permettant à la National Libfary of Canada to Bibliothèque nationale du Canada de reproduce, loan, distnibute or sel1 reproduire, prêter, distribuer ou copies of this thesis in microform, vendre des copies de cette thèse sous paper or electronic formats. la forme de microfiche/lnlm, de
reproduction sur papier ou sur format électronique.
The author retains ownership of the L'auteur conserve la propriété du copyright in this thesis. Neither the droit d'auteur qui protège cette thèse. thesis nor substantial extracts fiom it Ni la thèse ni des extraits substantiels may be printed or otherwise de celle-ci ne doivent être imprimés reproduced without the author's ou autrement reproduits sans son permission. autorisation,

Les composes naturels ont traditionnellement été la pierre angulaire de la découverte de nouveaux médicaments. En raison de la surexploitation des
ressources terrestres, l'attention se porte de plus en plus vers l'environnement marin comme source de nouveaux métabolites bio-actifs. Le squelette tétrahydrofurane semble assez répandu comme unité de base de nombreux composés naturels, tant d'origine terrestre que marine. De nombreux groupes de recherche se sont intéressés a leurs propriétés biologiques importantes ainsi qu'à la diversité des substituants sur le noyau tétrahydrofurane. Nous présentons une nouvelle stratégie permettant de construire une unité tétrahydrofurane 2,54ialkyl-3oxysubstituée qui sera appliquée à la synthese énantiosélective du (-)-trans-kumausyne et adaptée à celle du plakortone D. Également, nous proposons une approche à la synthese de plusieurs métabolites des algues brunes Notheia anomala.

RÉSUMÉ LONG
Le quart des médicaments utilisés à l'échelle mondiale provient de sources naturelles, principalement de microorganismes et de plantes
tropicales. En raison de la surexploitation des ressources terrestres, l'attention se porte de plus en plus vers l'environnement marin comme source de
nouveaux métabolites bio-actifs. Les composés isolés d'organismes marins possèdent des structures variées et parfois fort complexes, et pour beaucoup d'entre eux, il n'existe pas d'homologues dans le monde terrestre. L'étude d'une telle variété de composés représente un intérêt certain et un défi considérable pour le chimiste. La séparation de ces composés et l'élucidation de leur structure exigent le recours à l'arsenal complet des méthodes de séparation et d'analyse. Leurs synthèses sont souvent de parfaits chef-
d'oeuvres et, tout comme pour les composés d'origine terrestre, un grand
nombre d'analogues peuvent être imaginés, synthétisés et testés pour diverses activités pharmacologiques.
Le squelette tétrahydrofurane semble assez répandu comme unité de
base de nombreux composés naturels, tant d'origine terrestre que marine.
Malgré les nombreux développements réalisés au cours des dernières années dans ce domaine, la synthèse de ces unités polysubstituées demeure un sujet de recherche actuel, tant dans le milieu académique qu'industriel. Ces composés offrent un terrain fertile pour la mise au point de méthodes efficaces de préparation stéréocontrôlée de ces unités tétrahydrofuranes substituées.
Les travaux présentés dans cette thèse portent sur la synthèse de composés naturels d'origine marine possédant une unité tétrahydrofurane. En
particulier, nous avons mis au point une stratégie générale nous permettant de construire une unité tétrahydrofurane 2,5-dialkyl-3-oxysubstituée, et ce de

iii
manière concise et hautement efficace. Notre stratégie repose sur une réaction d'alkoxycarbonylation-factonisation catalysée par le Pd(ll). Elle sera appliquée a la synthèse énantiosélective du (-)-tram-kumausyne et adaptée à celle du plakortone 0. Également, nous proposons une approche a la synthèse de plusieurs métabolites des algues brunes Notheia anomala.
II est à souligner que notre stratégie offre une grande flexibilité quant à la manipulation de la stéréochimie et des divers substituants. Ceci ouvre donc la voie à la préparation d'autres composés naturels, et laisse place à la création de nombreux analogues synthétiques.

REMERCIEMENTS
Je voudrais tout d'abord remercier le professeur John Boukouvalas de
m'avoir accueillie dans son laboratoire, me permettant ainsi de parfaire mon apprentissage d'une discipline scientifique des plus stimulantes.
Je désire aussi remercier mes collègues de laboratoire, anciens et présents, qui ont su créer une atmosphère de travail agréable et tisser une complicité à toute épreuve face aux aléas du métier.
Mes remerciements s'adressent aussi à messieurs Jean Laferrière, Pierre Audet, Jocelyn Trernblay et Martin Plante qui m'ont apporté un soutien technique grandement apprécié.
Mes remerciements les plus sincères à ma famille et mes ami(e)s qui m'ont apporté leurs encouragements et leur support tout au long de mes études.

TABLE DES MATIÈRES
* RESUME COURT ..................................................................................................... i RESUME LONG ....................................................................................................... i i REMERCIEMENTS.. ............................................................................................... iv
....................................................................................... TABLE DES MATIÈRES v . . ...................... .............................. LISTE DES TABLEAUX ET SCHÉMAS ... XII
........................................................ NUMÉROTATION ET ABRÉVIATIONS xiv INTRODUCTION ...................................................................................................... 1
1. Les océans: source de composés nouveaux aux propriétés prometteuses. ......................................................................................... 1
.................. 2. Structures et activités des composés d'origine marine 4 3. Méthodes de préparation de tétrahydrofuranes substitués.. ........ -9 4. Notions de chimie du palladium ....................................................... 12
.......................................................... 5. Exposé du sujet de recherche .17
PARTIE THEORIQUE
CHAPITRE 1: SYNTHÈSE TOTALE DU (-)-TRANS-KUMAUSYN E
. . 1. Ongine du composé ........................................................................... 23 2. Autres synthèses du composé. ........................................................ -25

. . ................................................................... . 3 Analyse ktrosynthet~que 31
................................... 4 . Première voie de synthèse du 1 , 3-di01 anfi 32 ................................. . 5 Deuxième voie de synthèse du 1.3diol anti -39
..................................... ................. 6 . Phase finale de la synthèse ... 42
CHAPITRE 2: VERS LA SYNTHÈSE TOTALE D'UN MÉTABOLITE DES ALGUES BRUNES NOTHEIA ANOMALA
. . . 1 Origine du composé ........................................................................... 50
2 . Autres synthèses du composé ......................................................... -51 . . .............................................. 3 . Analyse rétrosynthettque .............. .. -60
4 . Première voie de synthèse du 1, 3diol syn .................................... 61 5 . Deuxièmevoiedesynthesedu1, 3diolsyn .................................. 62
.............................................................. 6 . Phase finale de la synthèse 63
CHAPITRE 3: SYNTHÈSE TOTALE DU PLAKORTONE D
. . ........................................................................... 1 . Origine du composé 69 .................................................. 2 . Analyse rétrosynthétique générale 72
3 . Approche à la synthèse de l'unité bicyclique ................................ 73 .............................................................................. 3.1 Rétrosynthèse 73
3.2 Vers la synthèse du 1, 3diol syn .............................................. -74
4 . Synthèse de l'unité bicyclique .......................................................... 80 .............................................................................. 4.1 Rétrosynthèse 80
....................... 4.2 Préparation d'une y-butyrolactone substituée 81
................... 4.21 Amplification de I'énantiosélectivité initiale 87
........................................................... 4.3 Synthèse du 1 , Mi01 syn 88
4.4 Synthèse du bicycle et preuve de stéréochimie ................... -91
4.5 Voie alternative de préparation du 1, 3diol syn .................... -93 ....................................................... . 5 Synthèse de la chaîne latérale 95
.................................................................... . 6 Couplage de type Wmig -97


viii
1.1 O (1 R,3RS,5R,7R)-2,6-dioxa-7-(t-butyldiphénylsilyloxy méthyl)-bicyclo [3.3.0] octan-3-01 59 ............................................ 1 1 9
1 .11 Oléfination du ladol 59 .............................................................. 1 2 1 1 -1 1 1 (2R13R, SR)-3-hydroxy-5-(t-butyldiphénylsiîyioxy-
méthyl)-2-(2(E)-5-triméthylsilyl-2-pentéynyI)- tétrahydrofurane 60.. ...................................................... .12 1
1.1 12 (2R13R,5R)-3-hydroxy-5-(t-butyldiphénylsilyloxy- méthyl)-2-(2(Z)-5-triméthylsilyl-2-pentèn4-ynyl)- tétrahydrofurane 61.. ......................................................... -123
1 -12 (2R13R,5R)-3-acétoxy-5-(t-butyldiphénylsilylo~méthyl)- 2-(2(E)-5-triméthylsilyl-2-pentèn-4-ynyl)-tetrahydro-
..... ....................... furane 62. ,.,. ... ....................................................... 1 24 1.1 3 (2R. 3R, 5R)-3-acétoxy-5-hydroxyméthyl-2-(2(E)-penten-
4-ynyl)-tétrahydrofurane 63.. ............................... .A 1.14 (2R,3Rl5R~-3-acétoxy-5-fomyl-2-(2(E)-penten4-ynyl)-
....................................................................... tétrahydrofurane 64.. 1 27 ....................................................... 1 .15 3-(triméthy1silyl)-1 -pentene 65 129
1 .16 (2Rl3R,5R)-3-acétoxy-5-(1 (S)-hydroxy-3(E)-hexeny1)- ............................. 2-(2(E)-penten-4-ynyI)-tétrahydrofurane 66.. -1 30
................................................................. 1 .17 (-)-trans-kumausyne 35 1 32

2.6 (2s. 3s. SR)-3-hydroxy-2-((2EZ)pentène)-5-(t-butyldi- phénylsilyloxyrnéthyl)-tétrahydrofurane 83 .............................. -140
2.7 (2S, 3s. SR)-2-hydroxy-3-pentyI-5-(f-butyldiphénylsilyl- oxyméthy1)-tétrahydrofurane 84 ..................... .... ...................... 142
2.8 (2s . 35. 5R)-3-acétoxy-2-pentyl-5-(f-buty1diphénylsilyl- .................................................... oxyméthy 1)-tétrahydrohirane û5 143
2.9 (2S, 3s. 5R)-3-acétoxy-2pentyl-5hydroxyméthyl-tétra- ................................................................................. hydrofurane 86 145
2.10 8rionyn-141 88 ................................................................................ 146 ............................................................................... 2.1 1 8-nonèn-141 89 -147
..................................................................... 2.12 9-bromo-1 nonène 90 148
CHAPITRE 3 ......................................................................................................... 150
.............................................. Alkylation de la butyrolactone 102 150
3.1 1 (3R. 5S)4,5.dihydro3éthyl.5.(f.butyldiphény 1.
silyloxyméthyl)-2(3H)-furanone 103.. ............................. 150
3.12 ( 3 5 5S)4,5.dihydro3éthyl.5.(t.butyldiphény 1. silyloxyméthy1)-2(3H)-furanone 104.. ............................. 151
Hydroxylation des butyrolactones 103 et 104 ........................... 153
3.21 (3S, 5S)-4.5-dihydro-3-hydroxy-3-6thyl-5- (f-butyldiphénylsilyloxyméthyl)-2(3H)- furanone 105 ....................................................................... 153
3.22 (3R, 5S)4,5.dihydro.3hydroxy.3-éthyt. 5. (t-buty ldiphénylsilyloxyméthyl)-2(3H)-
...................................................................... furanone 106 -155
(2RS, 3s 5s j~2,3~ciihydro~y~3éthyl~5~(t~butyldiphény 1. .......................................... silyloxyméthy 1)-tétrahydrofurane 107 -156
(1 S, 3s)-1 ,34hydrox y.1 -(t-butyldiphénylsilyloxy- méthy1)-Sthyl4pentene 108 ...................................................... 158 (4S)4-hydroxy-4éthyI-1 -(t-butyldiphény1silyloxy)-5-
......................................... ................................ hexèn-2ane 'i 10 ,... 159
(4s)-4-(triméthylsilyloxy)-4éthyl-1 -(t-butyldiphénylsilyl- ................................................................ oxy)-54exèn-2+ne 1 14 -161



LISTE DES TABLEAUX ET SCHÉMAS
Tableau I
TABLEAU
................................. Sélectivité des essais d'hydroxylation 85
Schéma 1 . C
Procede Wacker ....................................................................... 12
...................... Schdma 2 Intermédiaires formés par le Pd ... ................ 13 ........................................ Schéma 3 Oxydation d'alcènes avec le Pd(ll) 14
............................................................................ Sc héma 4 Carbonylation 15 . . Schéma 5 Strategiedesynthèse .............................................................. 19
Schéma 6 Plan général de synthèse pour les composés isolés ................................................................................. des algues 20
Schéma 7 Plan général de synthèse pour le plakortone D ................. 21
Schéma 8 Exemples de composés isolés des espèces
Schéma Schéma Schéma Schéma Schéma Schéma Schéma Schéma Schéma
................................................................................... Laurencia 24
Composés isolés de L . nipponica Yamada ....................... -25 ....................................... Synthèse de Oveman (1 8 étapes) 26
Synthèse de Osumi et Sugimura (1 9 étapes) ..................... 27 Synthèse de Martin (22 étapes) ............................................ 29
................................................ Synthèse de Lee (23 étapes) -30
................................ Retrosynthèse du (-).trans.kurnausyne 31
Modes d'attaque du MoOPH .................................................. 35
Origine de la sélectivité avec le MoOPH .............................. 36 ...................................... Mode d'attaque du réactif de Davis 37


Tout au long de ce travail, les composes chimiques seront nommés, pour la plupart, en ne mentionnant que les fonctions importantes et ils seront
désignés par des chiffres arabes en caractère gras. Ainsi, le composé 52 sera nommé lactol.
Les références bibliographiques seront représentées par des chiffres
arabes placés en exposant. De plus. ies abréviations suivantes seront utilisées afin d'alléger le texte:
Ac:
AlBN: BTAF: DEAD: DIBAH: DMAP: DME: DM F: DMSO:
Éq. : HMDS:
CH3CO- azo-bis-(iso-butyronitrile) fluorure de benzyltriméthylammoniurn
diéth ylazodicarboxylate hydrure de di-(iso -butyl)aluminium 4-diméthylam inopyridine 1,2-diméthoxyéthane N, N'diméthylfomam ide diméthylsulfoxyde équivalent molaire 1.1 ,1,3,3.3-hexaméthyldisilazane

HMPA: HPLC: IC: IE: IR KHMDS: LDso: LD A: LiHMDS:
KAPA MoOPH:
MS:
MsCI: NaHMDS:
NBS:
NOE: P.&: P-f.:
PG: P y: PTSA:
RMN: TBAF: TBCD: TBDPS: TBHP:
TESOTf
THF: TMEDA: TMS :
TMSOTt
hexaméthylphosphoramide
chromatographie liquide haute pression ionisation chimique
ionisation électronique spectroscopie infra-rouge
sel de potassium de HMDS
dose léthale pour 50% des animaux du groupe testé
di-(iso-propylam ine) de lith ium sel de lithium de HMDS 3-arninopropylamidure de potassium
oxodiperoxyrnolybdenum(pyridine)hexaméthyl- phophoramide spectroscopie de masse
chlorure de méthanesulfonyle
sel de sodium de HMDS
N-bromosuccinimide
effet Overhauser nucléaire (en RMN) point d'ébullition point de fusion
groupement protecteur
py ridine acide p-toluènesulfonique spectroscopie de résonance magnétique nucléaire fluorure de tetrabutylammonium
2,4,4,6-tétrabromo-2,s-cyclohexadiènone t-butyldiphénylsilyle
hydroperoxyde de t-butyle
trifluorométhanesulfonate de triéthylsilyle tétrahydrofurane N, N, Nt, Nt-tétraméthy léthylenediam ine
triméthylsilyle
trifluorométhanesulfonate de triméthylsilyle


INTRODUCTION
Le quart des médicaments utilisés à l'échelle mondiale provient de sources naturelles, principalement de microorganismes et de plantes
tropicales. En raison de la surexploitation des ressources terrestres, l'attention se porte de plus en plus vers l'environnement marin comme source de
nouveaux métabolites bio-actifs. Les médicaments anti-cancer et anti-viraux sont des inhibiteurs de la croissance cellulaire qui doivent agir sélectivement
sans affecter l'hôte. Le cancer est la deuxième cause de décès dans le monde occidental et on estime que 60% de toutes les maladies des pays développés
sont une conséquence d'infections virales. Bien que les composés d'origine terrestre et leurs dérivés aient été exploités depuis fort !ongtemps, leur efficacité pour le traitement de nombreuses maladies régresse à mesure que les pathogènes développent des resistances à leur endroit. Par conséquent. malgré la disponibilité de médicaments pour traiter ces maladies, des composés plus efficaces deviennent nécessaires.
Les océans, berceau de la vie. couvrent les trois quarts de la surface de
notre planète. y abritent plus de 80% des organismes vivants et pourtant. restent la ressource naturelle la moins exploitée. Les milliers d'espèces végétales et animales habitant les mers produisent une variété de métabolites beaucoup plus grande que les espèces terrestres. Certains, tels les alginates, la caragénine ou l'huile de foie de morue sont utilisés depuis fort longtemps par l'Homme et contribuent à son alimentation ainsi qu'à sa santé. Également, la
pharmacopée chinoise ancienne recommandait l'usage de certaines algues pour guérir diverses maladies telles que le goitre, les désordres gastro-

intestinaux ou contenaient des
les abcès. L'expérience avait démontré que ces algues principes actifs dont on ignorait évidemment la nature.
D'autres composés, par contre, sont des toxines que la plante ou l'animal
synthétise et qui jouent un rôle important dans la vie marine: elles peuvent éloigner un prédateur ou au contraire attirer une proie, déclencher une attraction sexuelle, servir de moyen de défense, inhiber la croissance d'une espèce concurrente ou ennemie, ou à l'opposé, favoriser la croissance en symbiose de certaines espèces, etc.. Ces toxines, notamment, ont été beaucoup étudiées car la toxicité indique une activité physiologique potentielle, donc la possibilité d'obtenir un médicament.
La vie, dans son expression la plus simple, est apparue en premier dans les mers, et il semble que la Nature ait tenté diverses expériences avant que des organismes plus évolués ne fassent leur apparition sur la terra ferma. L'étude d'organismes primitifs, quasi auto-suffisants, a conduit à l'isolation d'une grande variété de composés chimiques, provenant de leur métabolisme. Les animaux évolués cependant, dépendent les uns des autres pour les éléments dont ils ont besoin pour supporter leur mode de vie complexe. De ce point de vue, il est donc intéressant d'étudier en détail les éponges et les coélentérés qui sont de véritables fossiles vivants, ayant très peu évolués depuis des centaines de milliers d'années.
De nombreux produits isolés de plantes et d'animaux d'origine marine ont été caractérisés et démontrent un potentiel intéressant pour le traitement de nombreuses maladies. On y retrouve des antibiotiques, des antiviraux, des antitumoraux, des anticoagulants, des hémolytiques, des stimulants, des dépresseurs, des fongicides, des insecticides, etc ... L'étude d'une telle variété de composés représente un intérêt certain et un défi considérable pour le chimiste. L'isolation de ces composés et l'élucidation de leur structure exigent le recours à l'arsenal complet des méthodes de séparation et d'analyse. Leurs synthèses sont souvent de parfaits chef-d'oeuvres et, tout comme pour les composés d'origine terrestre, un grand nombre d'analogues peuvent être imaginés, synthétisés et testés pour diverses activités phamacologiques. Ces produits naturels peuvent donc entrer directement dans la pharmacopée moderne ou servir de modèles pour la synthèse de médicaments nouveaux et

efficaces. A titre d'exemple, le composé manoalide 1, isolé d'une éponge du
Pacifique,' a engendré à lui seul plus de 300 analogues synthétiques, dont un nombre significatif se sont retrouvés en essais cliniques comme agents anti-
inflammatoires.
Les travaux pionniers de W. Bergmann et E. Lederer, dans les années 30,
visaient essentiellement à étendre la "Chimie des Substances Naturelles" au
monde marin. C'est dans les années 70 que la chimie organique marine, fondamentalement interdisciplinaire, se reconnaît comme spécialité à l'intérieur
du vaste domaine des "Substances Naturelles". La publication en 1973 de
l'ouvrage de P.J. Scheuerz "Chemistry of Marine Natural Products" et la tenue a Aberdeen en septembre 1975 du premier "International Symposium on Marine
Natural Products" ont été les premières manifestations de l'intérêt et l'importance de cette nouvelle discipline. Depuis, le nombre annuel de
publications touchant tous les aspects de la synthèse de composés d'origine marine s'est accru rapidement. Les ouvrages de P.J. Scheuer3-9 et les revues
annuelles de la littérature de D. J. Faulknerl o-22 s'avèrent des références indispensables. De nos jours, ce domaine de recherche est plus que jamais
florissant et implique des chimistes organiciens, des biologistes marins, des pharmacologistes, des microbiologistes et des ethnologues.
Les composés naturels ont traditionnellement été la pierre angulaire de la découverte de nouveaux médicaments. Malgré les progrès remarquables en
synthèse organique, l'avènement de la chimie combinatoire, l'utilisation de la modélisation moléculaire et l'élucidation de nombreux mécanismes d'action, on
est encore loin d'une approche totalement rationnelle au développement de

nouveaux médicaments. L'isolation de produits naturels à partir de nouvelles sources continuera d'être une activité essentielle pour fournir aux chimistes de
synthèse de nouveaux composes qui les mèneront sur la piste de médicaments potentiels. D'ailleurs, de nombreuses compagnies pharmaceutiques ont établi
au cours des dernières années des alliances stratégiques avec des laboratoires académiques afin d'établir des programmes de recherche visant la découverte et i'exploitation de nouveaux composés d'origine marine.
0 0 - 1 . - 2. Structyyes et acvvites des composés d origine rnpLjpe
Les composes isolés d'organismes marins possèdent des structures
variées et parfois fort complexes, et pour beaucoup d'entre eux, il n'existe pas
d'homologues dans le monde terrestre.

A ce titre, la maitotoxine 2, isolée des dinoflagelles Gambierdiscus toxicus, a suscité beaucoup d'attention23 pour les raisons suivantes:
cette molécule dont le poids moléculaire est de 3422 Da (sous forme de sel disodique), excède celui de n'importe quel autre composé naturel connu, à l'exception des biopolymeres. La détermination de
sa structure a représenté un défi de taille;
elle joue fort probablement un rôle dans les intoxications chez l'Hom- me par certaines espèces marines et également dans la mort mas- sive et subite de poissons (marée rouge). L'élucidation de son méca-
nisme d'action revêt une grande importance dû aux implications socio-économiques engendrées par ces intoxications mais aussi pour la mise au point de contre-mesures;
La DL50 chez les souris (50 nglkg) en fait la plus puissante toxine non-peptidique connue, surclassant la palytoxine 1 2, une autre toxine puissante dont la synthese totale a été complétée par le groupe de Kishi24 en 1994.
En 1997, Nicolaou et collaborateurs25 ont réalisé la première synthese du eleutherobin 3 et ont ainsi déterminé sa stéréochimie absolue. Ce composé,
isolé d'un corail du Pacifique (près des côtes d'Australie), possède des propriétés anti-tumorales et on va même jusqu'à le qualifier de 'Yaxol des mers" puisqu'il démontre un mécanisme d'action similaire à ce dernier.26 La synthèse du eleutherobin vient tout d'abord pallier au problème d'obtenir une quantité adéquate de ce produit naturel peu abondant pour des tests visant a déterminer son potentiel thérapeutique, et elle ouvre la voie à la création de nombreux analogues. Initialement, les chercheurs qui ont isolé le composé disposaient de 10 mg de eleutherobin, soit l'approvisionnement mondial des trois années précédentes! Même si les sources peuvent facilement être localisées, on doit tenir compte des dommages potentiels qu'engendrerait une récolte massive de spécimens. La synthèse chimique vient donc A la rescousse en permettant d'obtenir des quantités suffisantes pour étudier tout le potentiel de ces composés. Parfois, on ne rapporte aucune activité pour un

nouveau composé tout simplement parce que les quantités disponibles pour
réaliser les tests ne sont pas suffisantes.
a-3) lu- -
Une telle complexité de structure n'est cependant pas la règle. On retrouve dans le monde marin des composés aussi simples que des alcanes
halogénés. La découverte que de nombreux composés d'origine marine comportaient des halogènes a suscité un vif intérêt étant donné que l'inclusion
de ces atomes dans la biosynthèse de ces produits semblait plutôt inusitée. Maintenant, il est reconnu que ces composés ne sont pas le résultat de la
présence de polluants générés par l'Homme mais qu'ils sont partie intégrante de notre écosystème depuis la nuit des temps.Z7 En effet, l'eau de mer est riche en halogènes, soit CI- 19.000 mg / L, B r 65 mg 1 L et 1- 1 103- 5x1 04 mg 1 L. ce qui permet aux organismes marins d'incorporer ces éléments ( B r étant au premier rang) dans des structures organiques covalentes. Pour les organismes terrestres, cette incorporation est un processus plutôt rare. Une corrélation directe a été établie entre la présence de composés halogénés et l'activité antimicrobienne dans plus de 1200 organismes marins. II serait donc probable que l'on voie, dans un proche futur, le développement de composés halogénés d'une grande importance pour leurs applications phamaceutiques.2*
Le squelette tétrahydrofurane semble assez répandu comme unité de
base de nombreux composés naturels, tant d'origine terrestre que marine.
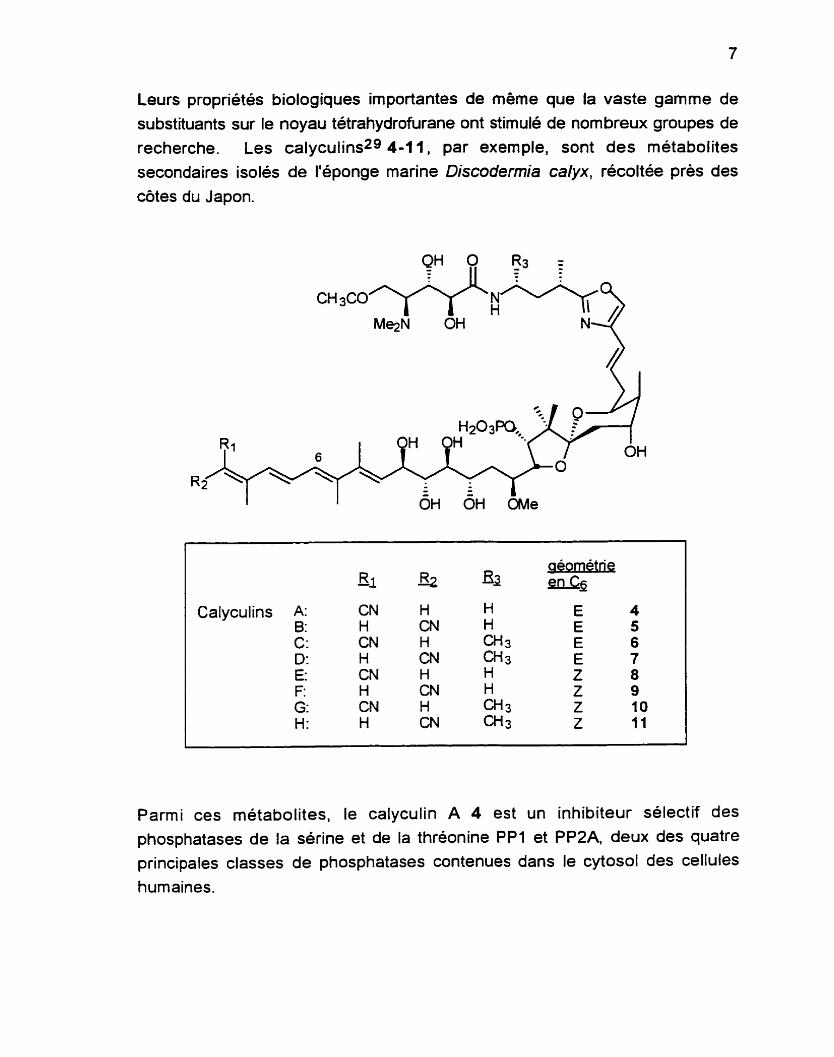
Leurs propriétés biologiques importantes de même que la vaste gamme de
substituants sur le noyau tétrahydrofurane ont stimulé de nombreux groupes de
recherche. Les calyculins29 4-1 1, par exemple, sont des métabolites secondaires isolés de l'éponge marine Discodermia calyx, récoltée près des
côtes du Japon.
Calyculins
Parmi ces métabolites, le calyculin A 4 est un inhibiteur sélectif des phosphatases de la sérine et de la thréonine PP1 et PPZA, deux des quatre principales classes de phosphatases contenues dans le cytosol des cellules humaines.
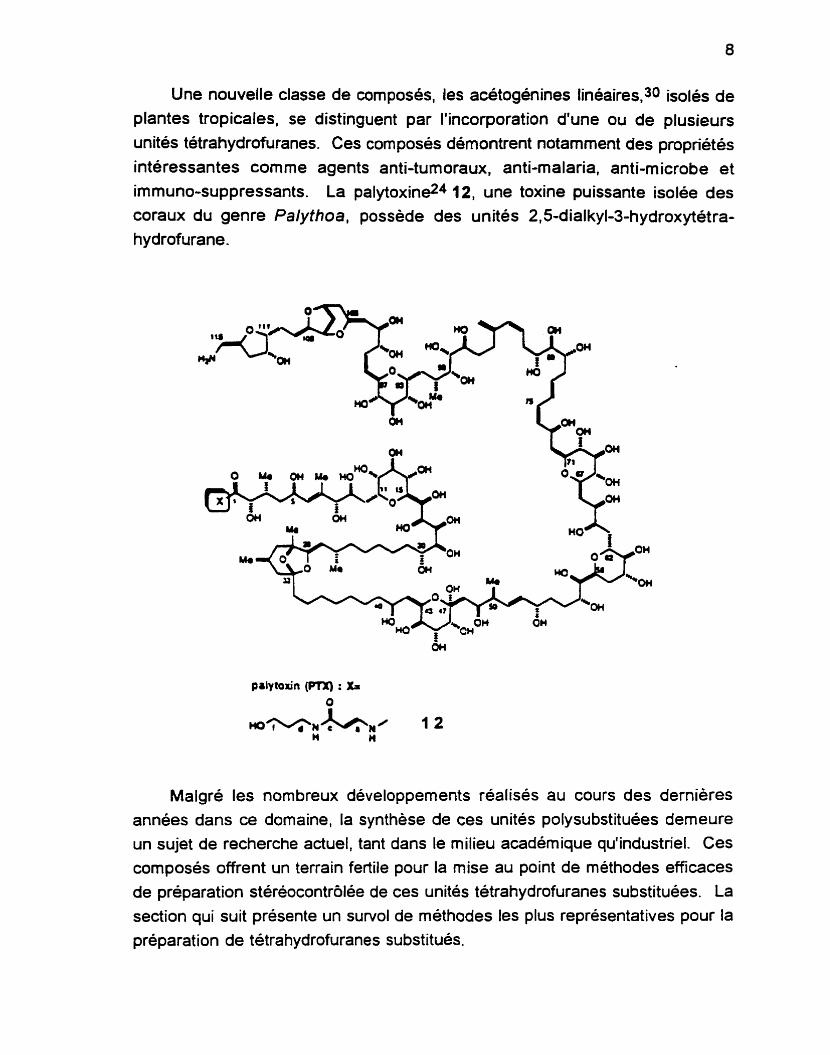
Une nouvelle classe de composés, les acétogénines linéaires,30 isolés de
plantes tropicales, se distinguent par l'incorporation d'une ou de plusieurs unités tétrahydrofuranes. Ces composés démontrent notamment des propriétés
intéressantes comme agents anti-tumoraux, anti-malaria, anti-microbe et immuno-suppressants. La palytoxine24 12, une toxine puissante isolée des coraux du genre Palythoa, possède des unités 2,5-dialkyl-3-hydroxytétra- hydrofurane.
Malgré les nombreux développements réalisés au cours des dernières années dans ce domaine, la synthèse de ces unités polysubstituées demeure un sujet de recherche actuel, tant dans le milieu académique qu'industriel. Ces
composés offrent un terrain fertile pour la mise au point de méthodes efficaces
de préparation stéréocontrôlée de ces unités tétrahydrofuranes substituées. La
section qui suit présente un survol de méthodes les plus représentatives pour la
préparation de tétrahydrofuranes substitués.

En particulier, de nombreuses approches ont été développées pour la
préparation de tétrahydrofuranes 2,5-disubstitués.31
La cyclisation d'alcools sur des alcènes est une des voies les plus directes pour la préparation de ces dérivés. Les règles de Baldwin32 stipulent que les cyclisations 5-exo sont particulièrement favorisées. II n'est donc pas surprenant, dans des cas d'iodoéthérification où il y a compétition entre le 5- exo et le Gendo, que ce soit le cycle à 5 membres qui prédomine. Cette méthode a été utilisée pour la synthèse de l'acide homonactique 15, dont le produit naturel tétranactin est un tétramère.33
Les cyclisat ions radicalaires sont parmi les méthodes les plus efficaces pour la formation de cycles à 5 membres. La majorité des cyclisations
radicalaires en chimie des hétérocycles ont été réalisées avec l'hydrure de
tributylétain (Bu3SnH) ou tout autre trialkylétain. Les conditions réactionnelles
sont généralement semblables, utilisant un léger excès de Bu3SnH avec une quantité moindre (1 0-25%) d'un initiateur radicalaire. Depuis quelques années, des exemples fort ingénieux ont été utilisés pour la synthèse
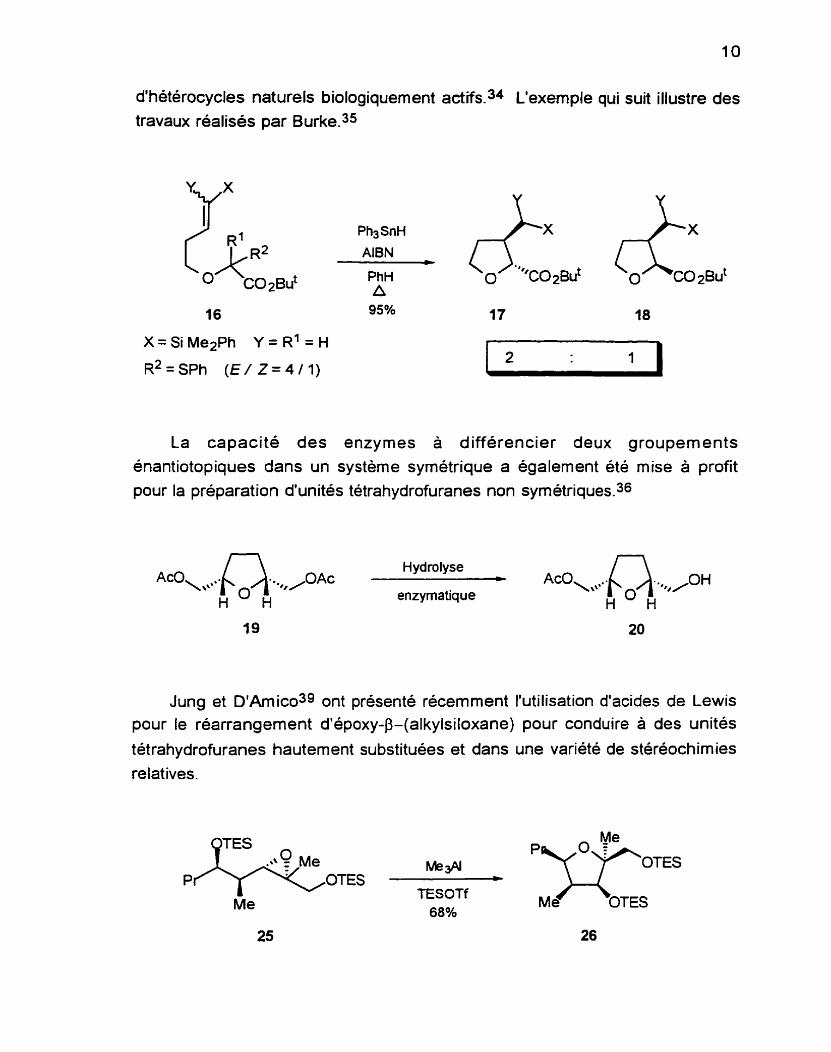
d'hétérocycles naturels biologiquement actifs.34 L'exemple qui suit illustre des
travaux réalisés par Burke?=
Ph3SnH
AlBN
P ~ H 0 "*CO~BU~ A
16 95% - ' 17
'1@ 48
La capacité des enzymes à différencier deux groupements
énantiotopiques dans un système symétrique a également été mise à profit pour la préparation d'unités tétrahydrofuranes non symétriques36
Hydrolyse 4
enzymatique
Jung et D'Amico39 ont présenté récemment l'utilisation d'acides de Lewis pour le réarrangement d'époxy-P-(alkylsiloxane) pour conduire à des unités
tétrahydrofuranes hautement substituées et dans une variété de stéréochimies relatives.
P OTES

On a également vu au cours des dernières années de nombreux travaux portant sur les méthodes de synthèse d'unités tétrahydrofuranes contiguës. L'ouverture d'époxydes3' (21 -22) . de même que la réaction multiple de
Williarnson3a (23 -24 ) ont été utilisées.
Une voie impliquant une catalyse par le Pd(ll) avec extension de chaîne par le CO a été mise au point.40
Cette méthode offre de grandes possibilités d'application en synthèse et ouvre la voie a d'autres développements. Nous présentons dans ce travail. une nouvelle méthode de préparation de tétrahydrofuranes trisubstitués qui repose sur la chimie du palladium. II s'avère donc important de discuter brièvement la
chimie de ce métal puisqu'elle représente l'étape-clé de chaque synthèse qui seront présentées ultérieurement.

4. Notions de c w i e du . .
L'utilisation du palladium comme catalyseur pour les hydrogénations et les oxydations remonte aux premiers temps de la chimie organique. Cependant, son développement a connu une croissance laborieuse. Alors que les premiers travaux n'examinaient que la coordination du palladium aux molécules organiques, la mise au point d'une synthese industrielle de
l'acétaldéhyde (procédé Wacker, 1953), faisant intervenir une oxydation de
l'éthylène par le Pd, a stimulé la recherche pour de nouveaux processus
catalysés par ce métal.
PdCI2+ H20 + H2C=CH2 - H3C-CHO + 2 HCI + Pd
Schéma 1: Procédé Wacker
Cet essor se reflète bien dans la parution de plusieurs ouvrages41-47 traitant du
grand potentiel de la chimie du palladium.
Le palladium offre beaucoup de possibilités pour la formation de liens carbone-carbone et, de tous les complexes de métaux utilisés comme catalyseurs en synthese, ceux du palladium demeurent en tête de liste pour
leur capacité d'adaptation et leur réactivité. La tolérance des réactifs de
palladium vis-à-vis de nombreux groupements fonctionnels, tels que carbonyle et hydroxyle, s'avère une caractéristique attrayante. Ainsi, les réactions
catalysées par le Pd peuvent être faites sans avoir à protéger ces derniers. Bien que les réactions impliquant le Pd devraient être réalisées sous
atmosphère inerte, les réactifs et catalyseurs du Pd ne sont pas particulièrement sensibles à l'oxygène et l'humidité, ni même aux acides. Bien
sûr, le palladium est un métal noble donc dispendieux, mais il l'est beaucoup moins que le Rh, le Pt ou le Os. Également, la toxicité du Pd n'a suscité aucun
problème jusqu'à ce jour. Plusieurs procédés industriels opérationnels (plus de dix) sont basés sur des réactions catalysées par le palladium.

les un
La réactivité des complexes du palladium dépend de son état d'oxydation,
deux plus communs étant O et +2. Fondamentalement, le Pd(ll) agit comme acide de Lewis alors que le Pd(0) est nucléophile. La chimie du palladium
repose sur la formation de deux types majeurs d'intemédiaires,47 le système x-
allyle (A) et l'espèce a-palladium (B) (Schéma 2).
WQ) q + - -Pd- X
A X = groupe partant
Ar- Métal py X = groupe partant
Schéma 2: Intermédiaires formés par le Pd
Pour les réactions catalysées par le Pd(O), le choix des substrats est habituellement réduit à ceux contenant une double liaison carbone-carbone et
un groupe partant quelconque (acétate d'allyle, halogénure d'aryle, triflate de vinyle,...). Le palladium sert alors de support activant la partie allyle, et fournit une charpente pour les réactions stéréocontrôlées.
Pd(PPh3 14
PPh .@lOEt -
THF, reflux *110Et
29 PhCH 2NHMe 30
87%

Si on se réfère au Schéma 2, les réactions catalysées par le Pd(ll) seraient potentiellement plus avantageuses puisque la nature d'oxydant du métal dans
cet état d'oxydation permettrait l'utilisation d'alcènes non fonctionnalisés. Étant réduit en Pd(0) lors de la réaction, on doit prévoir un moyen de regénérer le Pd(ll) pour rendre le processus catalytique. De fait, les catalyses impliquant le Pd(ll) ont longtemps été délaissées. Ce manque d'intérêt était en partie dû à la crainte d'une incompatibilité des substrats etbu produits multifonctionnalisés avec les conditions de réoxydation de même que le faible pourcentage de substrat transformé dans les cycles catalytiques.
Des développements dans les réactions impliquant la catalyse par le Pd(ll) ont permis de mettre au point la synthèse d'hétérocycles contenant des atomes d'oxygène, dans des wnditions douces. Un exemple typique de la chimie du Pd(l1) peut être illustré par l'oxydation d'alcènes (Schéma 3) dans laquelle un nucléophile tel que OH ou OAc attaque en premier lieu l'oléfine qui est complexée au métal, formant ainsi un intermédiaire dans lequel le métal se lie à la molécule organique par un lien a ("O-bonded-Pd(ll) intermediate"). La B- élimination subséquente de l'espèce Pd-H conduit au produit, et le Pd-H résultant se décompose pour donner Pd(0) et HX. Cette élimination résulte
habituellement en la formation de produits portant l'alcène dans la position la
plus stable. Cette réaction est appellée alkoxypalladation.
Schema 3: Oxydation d'alcènes avec le Pd(ll)
En principe, ces cyclisations sont rendues catalytiques par l'une ou l'autre des combinaisons de sels de Cu(l1) et 0 2 ou p-benzoquinone comme
réoxydants. Dans la majorité des cas, on utilise un excès de sel de Cu(ll), habituellement CU CI^, pour ne pas avoir à utiliser un deuxième oxydant dans le milieu réactionnel.

Mais ce qui demeure l'application la plus utile de ce type de réaction est
sans conteste la carbonylation, c'est-à-dire l'insertion de CO (Schéma 4) dans
les molécules organiques. L'intermédiaire de la réaction d'alkoxypalladation est trappe par le CO. Cette réaction est courante avec le palladium et certains autres métaux (Ni, Pt) et se déroule par migration du groupe alkyle du métal vers le carbone. La liaison x entre le Pd et le CO résulte d'un recouvrement des orbitales p anti-liantes vides du ligand par les orbitales dx du métal.
\ RPdX
HPdX Y
Schéma 4: Carbonylation
Les premiers travaux sur ces réactions n'impliquaient que des composés simples, des oléfines sans substituants (éthylène, cyclopentène). D'un point de
vue utilitaire, ces résultats étaient peu satisfaisants quant à leur application en synthèse mais ils ont ouvert la voie au développement de nouvelles méthodologies permettant d'effectuer la carbonylation sur des alcènes beaucoup plus complexes.

La version intramoléculaire de cette réaction, développée par Semrnelhack,48 est beaucoup plus efficace et donne accès a des synthons plus fonctionna1 isés et qui peuvent être modifiés de diverses manières.
5% PdCI2
R CO, MeOH C02Me
CuCh
II est également possible d'effectuer une double cyclisation en tandem, conduisant ainsi à une unité bicyclique ayant un potentiel fort intéressant du point de vue synthétique, bien que très peu d'applications existent à ce jour en synthèse de produits naturels.49
PdCI2, CU CI^
NaOAc, AcOH
Cette réaction, élaborée par Yoshida et collaborateurs,so~51 procède avec d'excellents rendements ainsi que des stéréosélectivités complètes. En effet, en raison de l'effet directeur du OH en C-3, seules les cis-lactones sont
observées. La seule limitation de cette réaction semble être la substitution sur la double liaison où de p!us faibles rendements ont été observés dans certains cas. Cette réaction est nommée alkoxycarbonylation-lactonisation. La réaction
peut s'appliquer à des systèmes 3-hydroxy-4-pentenylarnine (amino- carbonylation-lactonisation).s~2 On peut également obtenir des bis-lactones si le C-1 est un acide c a r b o x y l i q ~ e . ~ ~ ~ ~ ~
Elle se révèle un outil puissant pour bâtir un système complexe dans un contexte de minimisation des sous-produits de réaction ("atom-econorny").54

Améliorer l'efficacité de la synthèse de molécules organiques complexes constitue un des défis les plus stimulants pour le chimiste de synthese mais il ne suffit plus simplement d'améliorer la sélectivité des procédés. On cherche de nos jours à maximiser le nombre d'atomes de matériel de départ qui aboutit dans le produit final. D'un point de vue environnemental, la communauté chimique est consciente de la nécessite de réduire la quantité de déchets produits et la sur-consommation de matières premières- Bien entendu, certaines réactions telle que I'oléfination de Wittig s'avèrent difficiles à
abandonner puisqu'elles représentent une méthode efficace pour la formation de liens carbone-carbone, mais d'autres, tels que les couplages croisés, se révèlent des alternatives fort prometteuses. La recherche dans ce domaine est plus active que jamais.
5 . m o s é du suiet de recherchp
La planification d'une synthese, peu importe la complexité de la cible, commence tout d'abord par un processus d'analyse ré t rosynthét iq~e.~~ La
rétrosynthese s'avère être une technique pour transformer la structure d'une molécule donnée en une séquence de sous-structures progressivement plus simples. Ultimement, ce cheminement conduit à des composés disponibles commercialement. Le chimiste de synthèse cherche à identifier les liaisons
stratégiques qui pourraient être avantageusement clivées dans le processus de rétrosynthèse. Parallèlement, il cherche les réactions pour construire, dans le sens inverse, chaque lien brisé dans la rétrosynthese. La clé du succès à ce
stade est d'être très méthodique et de savoir découvrir les éléments subtils de la structure qui peuvent conduire à une stratégie à la fois efficace et élégante.
Une fois tous les schémas rétrosynthétiques envisagés ayant été explorés, le chimiste de synthese est maintenant en position d'évaluer la stratégie à adopter pour la construction de la moléculecible. II recherche des méthodes56 qui:
i) effectue une transformation sur une portion définie de la structure et pas une autre (chimiosélectivité);

i i)
iii)
iv)
oriente les partenaires d'une réadion dans un arrangement défini
(rég iosélect ivité);
crée l'orientation définie des divers éléments de la molécule les uns
par rapport aux autres (diastéréosélectivité);
permet la création d'une molécule chirale donnée sans présence de son image miroir (énantiosélectivité).
On cherche également à inclure, dans une stratégie bien planifiée, les
élém entss? suivants:
i)
ii)
iii)
iv)
v
v i)
vii)
réactions efficaces;
concision;
produits de départ facilement accessibles et peu dispendieux;
conditions de réactions pratiques;
flexibilité en cas de problèmes;
adaptabilité de la stratégie a d'autres composés de structure apparentée;
souci de l'environnement par rapport aux déchets générés;
et finalement, nouveauté, élégance et art!
De telles exigences représentent un défi stimulant. La synthèse est
devenue au cours des dernières années une force motrice pour l'avancement des sciences. Depuis les agents thérapeutiques et les polymères de haute
technologie jusqu'au développement de la nanotechnologie, la synthèse a laissé son empreinte sur notre santé, notre économie et notre mode de vie.
L'immense potentiel de la synthèse est loin d'avoir été entièrement exploité et

toutes les disciplines scientifiques bénéficieront encore des progrès futurs dans ce dornaine.58
Les travaux présentes dans cette thèse portent sur la synthèse de
composés naturels d'origine marine possédant une unité tétrahydrofurane. En particulier, nous avons mis au point une stratégie générale nous permettant de construire une unité tétrahydrofurane 2.5-dialkyl-3-oxysubstituée, et ce de manière concise et hautement efficace.
Notre stratégie repose sur la réaction d'alkoxycarbonylation-lactonisation catalysée par le Pd(ll) qui a été décrite auparavant. Avec ces unités bicycliques. nous avons imaginé pouvoir appliquer la séquence suivante? une réduction de la lactone, suivie d'une réaction de Wittig permet d'obtenir une unité tétrahydrofurane 2.5-dialkyl-3-oxysubstituée (Schéma 5), qui pourra être transformée ultérieurement. Plusieurs composés naturels provenant de sources diverses contiennent une telle unité. Nous allons présenter trois produits naturels pouvant être synthétisés à partir de cette approche.
Schéma 5: Stratégie de synthèse
Le Chapitre 1 présente la synthèse du (-)-trans-kumausyne60 35, un composé isolé des algues rouges du genre Laurencia.
Dans le Chapitre 2, on présente une approche à la synthèse du di0161 36,
un des nombreux métabolites des algues brunes Notheia anomala possédant une unité tétrahydrofurane. Cette stratégie permet aussi l'accès aux autres membres de cette famille ayant une unité tétrahydrofurane.
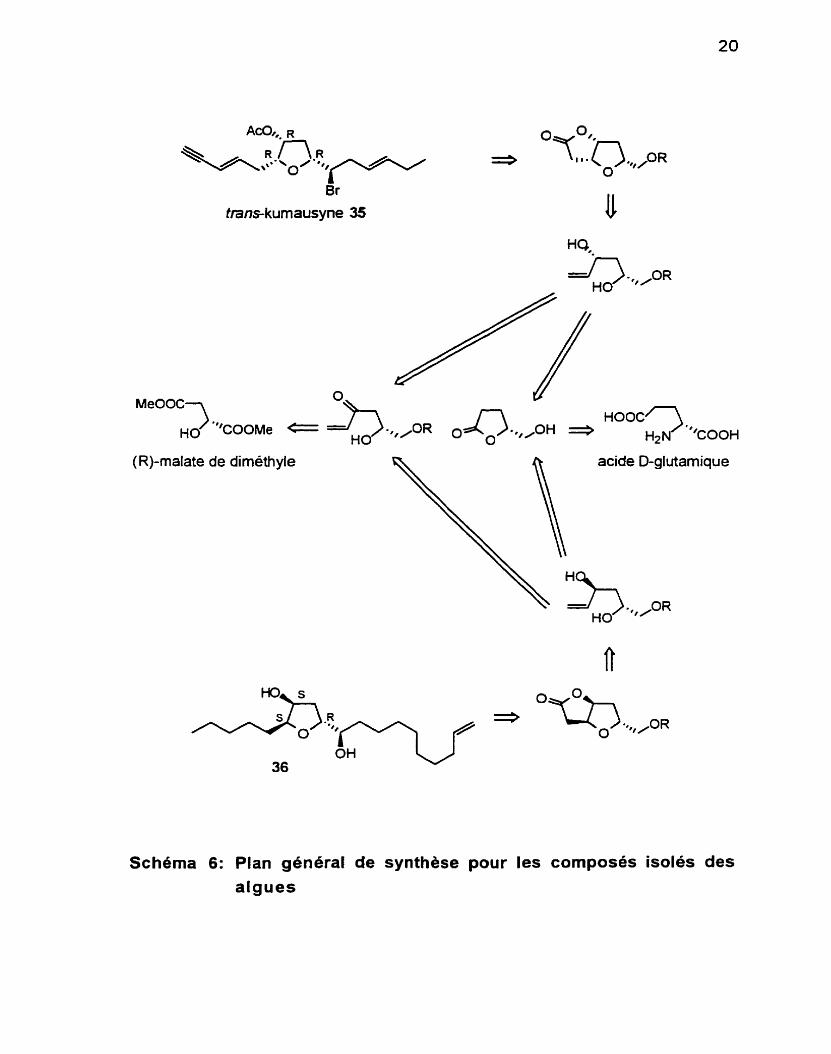
Schéma 6: Plan général de synthèse pour les composés isoles des algues

II est à souligner que notre stratégie offre une grande flexibilité (Sch6ma 6)
et qu'il nous est possible. B partir d'un même chiron de départ. d'accéder à l'une ou l'autre famille de tétrahydrofuranes. Ceci ouvre donc la voie à la préparation de nombreux composés naturels. et laisse place à la création de nombreux analogues synthétiques.
Également. nous mettrons à profit, dans le Chapitre 3, la réaction d'alkoxycarbonylation-lactonisation pour la préparation d'une unité bicyclique de type furane-furanone, qui sera utilisée dans la synthèse d'un nouveau produit d'origine marine, le plakortone DG2 3 7 (Schéma 7). Lors de l'isolation et de la caractérisation de ce composé, les chercheurs ont pu déterminer la stéréochimie relative de l'unité bicyclique mais celle du centre stéréogénique de la chaine latérale n'a pu être résolue.
plakortone D 37
Schéma 7: Plan général de synthèse pour le plakortone D

Nous allons donc prouver, par notre synthèse, la stéréochimie relative du substituant sur la chaîne latérale et par le fait même. la stéréochimie absolue du produit naturel.

CHAPITRE 1
Synthèse totale du (-)-trans-kumausyne
L'espèce marine Laurencia a été décrite pour la première fois en 181 3 par Lamouroux. On connaît maintenant qu'elle comprend une large variété
d'espèces d'algues que l'on retrouve à I'échelie de la L'étude des métabolites secondaires issus des algues rouges du genre Laurencia a véritablement débuté en 1953 par la parution d'un rapport de Obota et
F ~ k u s h i ~ ~ b sur l'huile essentielle de L. glandulifera. À ce moment, les méthodes d'analyse limitées avaient conduit ces auteurs à conclure que les
composants majeurs de cette huile étaient des sesquiterpenoïdes. Le groupe de lrie63c a ensuite, en 1965, isolé et déterminé la structure de laurencin 39, à partir de ces mêmes algues. Plusieurs groupes ont à leur tour suivi la voie
tracée par l'engouement des découvertes de lrie et de nos jours, les espèces Laurencia sont probablement les algues les plus étudiées. Plus de 250 produits naturels ont été isolés, plusieurs ayant des structures jamais rencontrées auparavant.
En particulier, des éthers cycliques comportant des atomes d'halogènes (Schéma 8), dont le squelette est composé de 15 atomes de carbone, sont des métabolites communs de plusieurs espèces de Laurencia. Ils se distinguent
par leur cycle oxane de diverses grandeurs. On présume qu'ils dérivent tous
de précurseurs acycl iq ues.63 dont I'orig ine peut être retracée jusqu'à l'acide hexadéca-4,7,10,13-tétraènoïque 42. L'isolation des laurédiols 41 (Schéma 8)
de L. nipponica est venue apporter une preuve pour valider ce postulat. On suppose qu'une cyclisation induite par un ion bromonium est à l'origine de la formation de ces éthers cycliques.
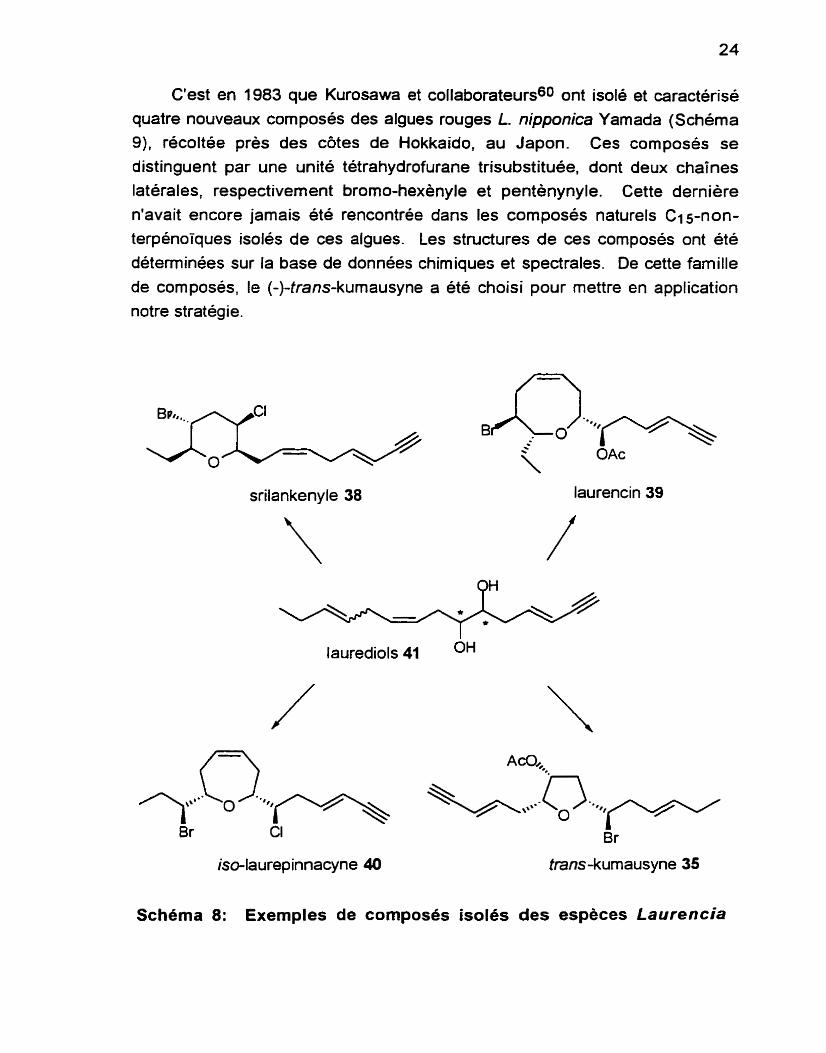
C'est en 1983 que Kurosawa et collaborateurs60 ont isolé et caractérisé quatre nouveaux composés des algues rouges L nipponica Yamada (Schéma
9). récoltée près des côtes de Hokkaido, au Japon. Ces composés se distinguent par une unité tétrahydrofurane trisubstituée, dont deux chaines
latérales, respectivement brorno-hexènyle et pentènynyle. Cette dernière n'avait encore jamais été rencontrée dans les composés naturels C15-non- terpénoiques isolés de ces algues. Les structures de ces composés ont été déterminées sur la base de données chimiques et spectrales. De cette famille de composés. le (-)-trans-kumausyne a été choisi pour mettre en application
notre stratégie.
srilankenyle 38 laurencin 39
laurediols 41 OH
iso-laurepinnacyne 40 trans-kumausyne 35
Schema 8: Exemples de composés isoles des espèces Laurencia

R = Ac, cis-kumausyne 43 R = Ac, trans-kumausyne 35
R = H. cisdésacétylkumausyne 44 R = H, tram-désacétylkumausyne 45
Scherna 9: Composés isolés de L. nipponica Yamada
À ce jour, trois synthèses du (-)-tram-kumausyne ainsi qu'une du (+)-
trans-désacétyl kumausyne ont été réalisées.
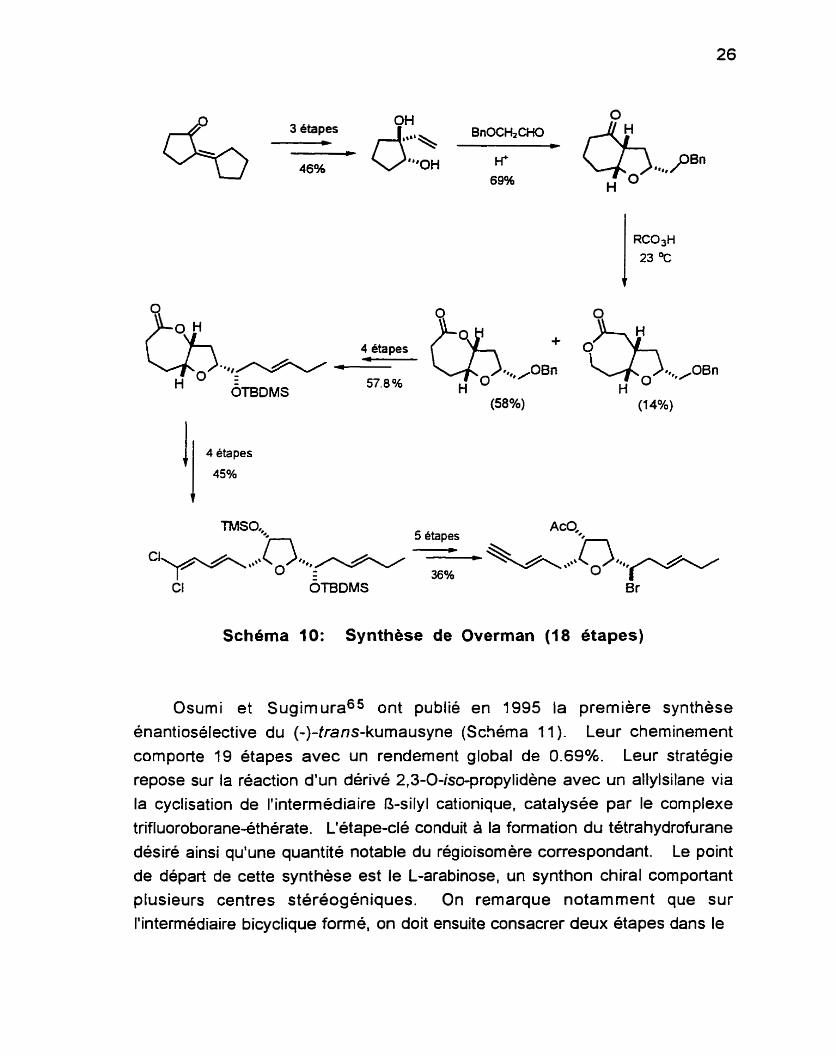
En 1991, Overman et collaborateurs64 (Schéma 10) ont préparé le (t)- t r an s-kumausyne via la formation stéréocontrôlée d'un dérivé
hydrobenzofuranone (préparé sous forme racémique à partir de composés disponibles commercialement) par un réarrangement Prins-pinacol. L'oxydation de ce dérivé avec l'acide m-chloroperbenzoique conduit a un mélange de régioisomères (4:l) pouvant être séparés par chromatographie. Cette synthèse comporte 18 étapes, avec un rendement global de 1.7%. Le
dérivé hydrobenzofuranone peut aussi être préparé en quelques étapes de
manière énantiosélective (84% ee) mais les auteurs n'ont pas jugé utile de
refaire la séquence pour obtenir le produit naturel.
4 étapes
45%
TMS O#,* AcO, 5 étapes *0
36 Or6 0
Schéma I O : Synthèse de Overman (18 étapes)
Osumi et sugimura65 ont publié en 1995 la première synthèse énantiosélective du (-)-trans-kumausyne (Schéma 11). Leur cheminement
comporte 19 étapes avec un rendement global de 0.69%. Leur stratégie
repose sur la réaction d'un dérivé 2,3-0-iso-propylidène avec un allylsilane via la cyclisation de l'intermédiaire R-silyl cationique, catalysée par le complexe trifluoroboraneéthérate. L'étape-clé conduit à la formation du tétrahydrofurane désiré ainsi qu'une quantité notable du régioisomère correspondant. Le point
de départ de cette synthèse est le L-arabinose, un synthon chiral comportant plusieurs centres stéréogéniques. On remarque notamment que sur l'intermédiaire bicyclique formé, on doit ensuite consacrer deux étapes dans le
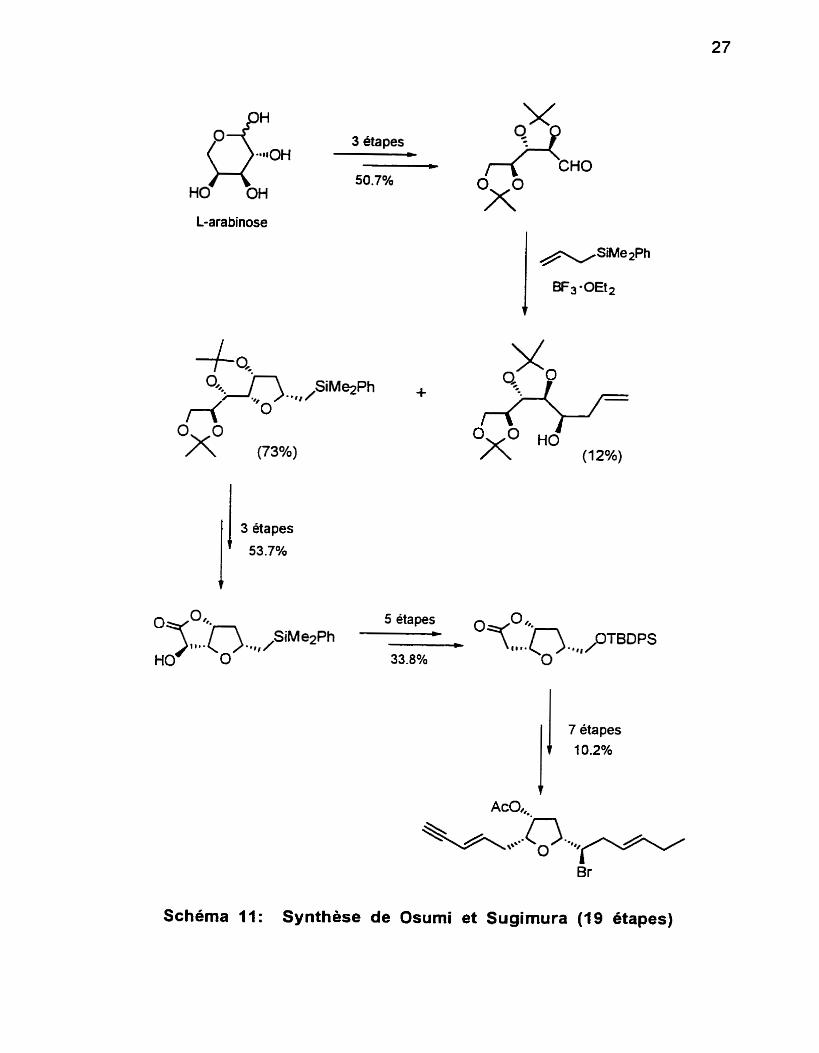
3 étapes 4 -
50.7Oh
3 étapes
53.7%
~$Q.,,,,siMe~ph
5 - étapes
HO 33.8%
l 7 étapes
10.2%
Schéma 11: Synthèse de Osumi et Sugimura (19 btapes)

seul but de cliver la fonction hydroxyle qui est absente dans le produit naturel mais qui s'avère indispensable pour cette stratégie. Paradoxalement, on doit également consacrer deux étapes pour transformer un lien C-Si en lien C-OH.
Au début de 1997, Martin et collaborateurs66 (Schéma 12) ont présenté
une synthèse du (+)-frans-désacétylkumausyne. Cette synthèse repose sur la cyclisation d'une chaîne hydroxy-alcène induite par un ion bromonium. Les auteurs ont ainsi tenté de mimer la voie proposée pour la biosynthèse de ce composé. La réaction conduit à un mélange 5:l d'un tétrahydrofurane intermédiaire dont la stéréochimie en C-3 doit être corrigée en fin de séquence. L'utilisation de l'époxy-alcool thréo permettrait d'accéder à l'unité
tétrahydrofurane avec la bonne stéréochimie pour les trois substituants. Cette voie a bien été tentée par les auteurs mais elle ne permet pas de sélectivité
(mélange 1 : 1 ) lors de la cyclisation induite par l'ion bromonium. Cette première
synthèse du (+)-trans-desacéty rendement global de 12.7%.
Tout récemment, Lee et col
kumausyne comporte 22 étapes avec un
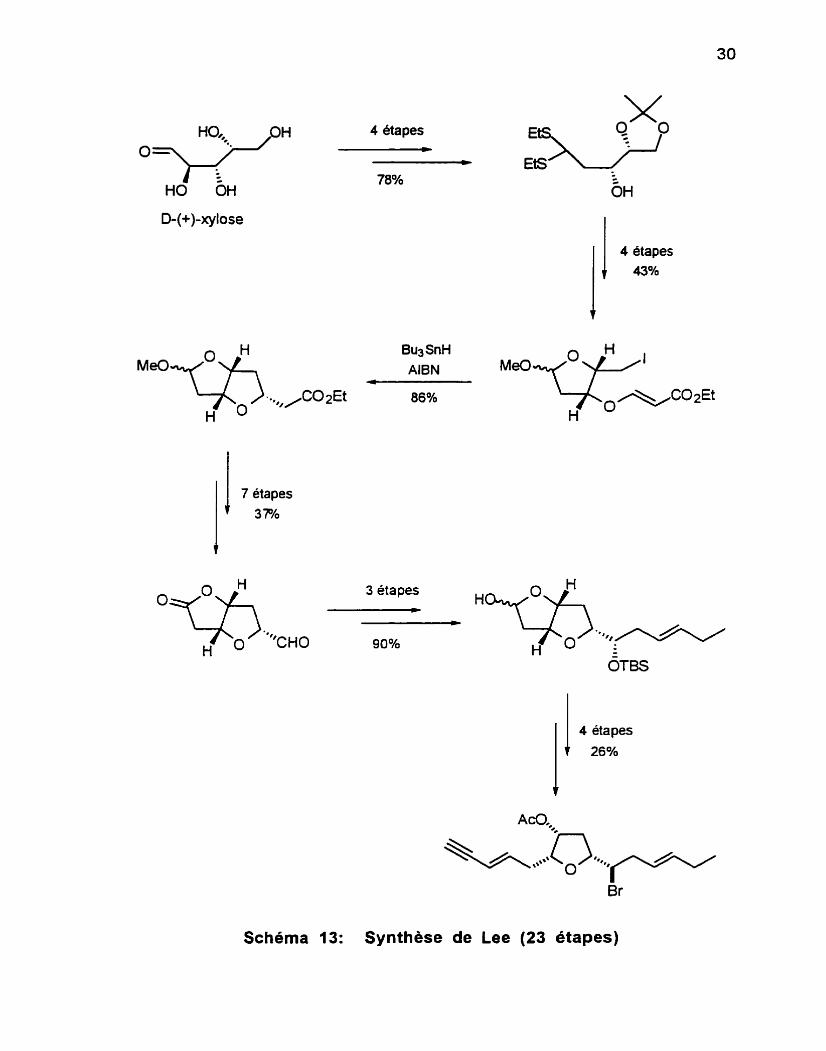
aborateurs67 (Schéma 13) ont mis à profit la cyclisation radicalaire de P-alkoxyacrylates pour la synthèse du (-)+ans- kumausyne. Cette réaction conduit à un seul et unique produit de cyclisation. Cependant, on doit convertir le composé en lactone car l'acétal cyclique obtenu ne résiste pas aux conditions pour le couplage avec un allylsilane. Ceci ajoute
donc des étapes supplémentaires. Cette séquence procède en 23 étapes avec un rendement global de 2.47%.
Plusieurs de ces synthèses ont en commun un intermédiaire bicyclique. Notre synthèse fait également appel à ce type d'intermédiaire et nous verrons que notre approche nous permet d'obtenir un bicycle en un nombre beaucoup plus restreint d'étapes. De plus, un seul et unique produit de cyclisation est
obtenu et ce, de manière hautement efficace.
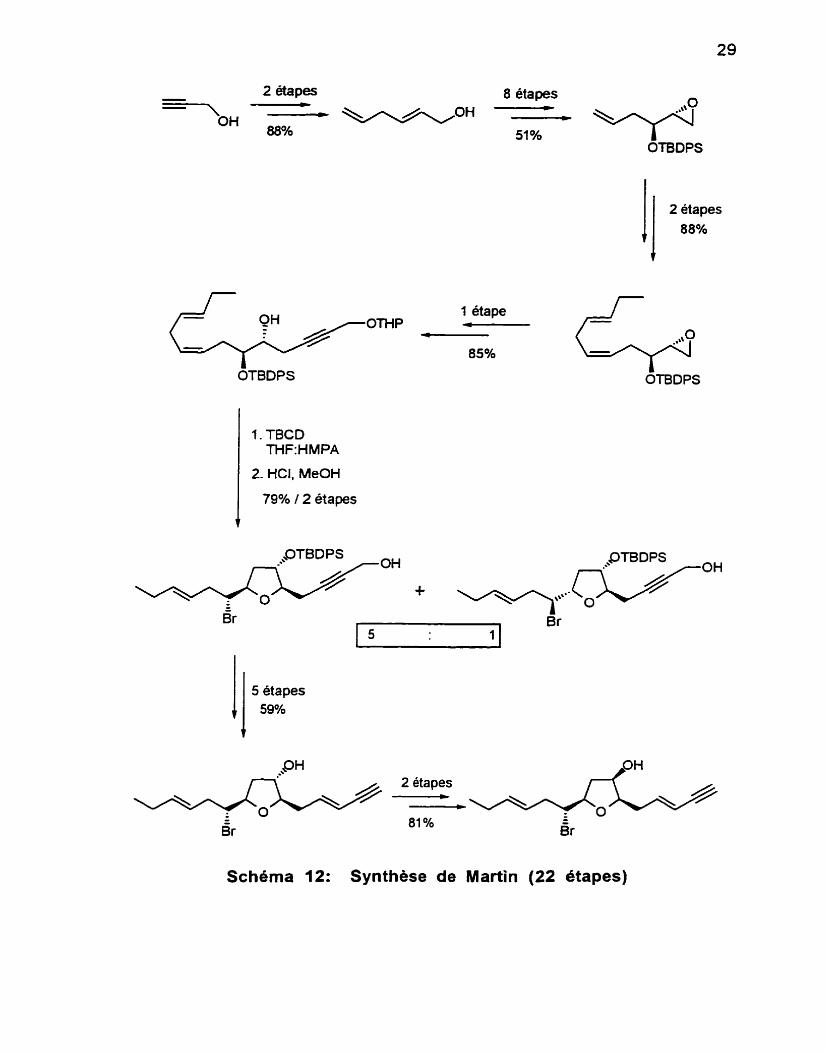
2 étapes 8 étapes =-, - - -OH --* OH 80% 51 %
OTBDPS
2 etapes 88%
1 2. HCI, MeOH
1 79% 1 2 étapes
1 étape - - 85% & -
OTBDPS
5 étapes 59%
PH b
2 étapes
&r 81 % er
Schéma 12: Synthèse de Martin (22 étapes)
4 étapes %
C
1! 4 étapes 43%
Bu3SnH AlBN
4
- C02Et "#,/
86% H M a 7 ~ ~ ~ ~ c o H
1 ' 7 étapes
3 Ph
3 étapes *
t
90%
OTBS
4 étapes
26%
Schéma 13: Synthèse de Lee (23 étapes)
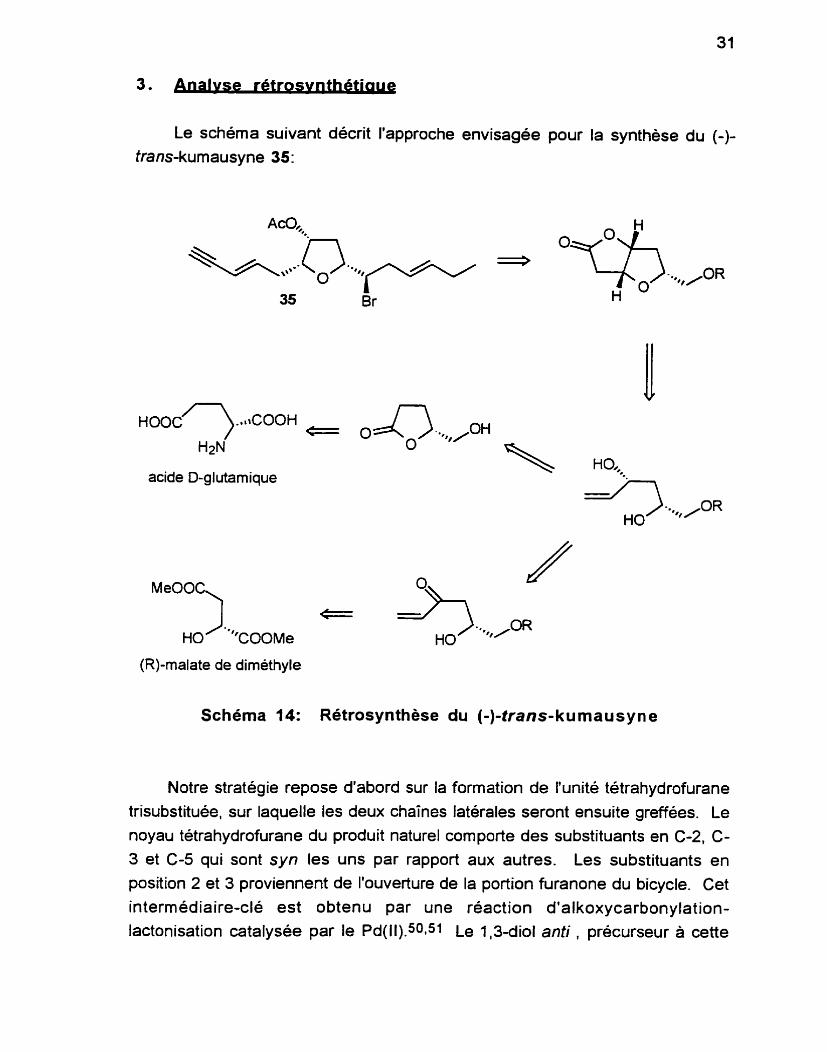
Le schéma suivant décrit l'approche envisagée pour la synthèse du (-)-
tram-kumausyne 35:
(R)-malate de diméthyle
Schéma 14: Rétrosynthèse du (-)-trans-kumausyne
Notre stratégie repose d'abord sur la formation de l'unité tétrahydrofurane trisubstituée, sur laquelle les deux chaînes latérales seront ensuite greffées. Le
noyau tétrahydrofurane du produit naturel comporte des substituants en C-2, C- 3 et C-5 qui sont syn les uns par rapport aux autres. Les substituants en position 2 et 3 proviennent de l'ouverture de la portion furanone du bicycle. Cet
intermédiaire-clé est obtenu par une réaction d'alkoxycarbonylation- lactonisation catalysée par le Pd(ll).501s1 Le 1,3-diol anti , précurseur à cette

transformation, peut être obtenu selon deux voies distinctes qui seront décrites. Le premier centre stéréogénique provient d'un synthon chiral commercialement disponible. Tous les autres centres seront élaborés à partir de la stéréochimie de ce dernier.
La désamination nitreuse de l'acide D-glutamique68 en solution aqueuse conduit à la lactone-acide 46, après cristallisation dans un mélange Ac0Et:benzène:éther de pétrole 35-60 O C . II est recommandé de purifier ce composé par recristallisation et non pas par distillation afin d'éviter la racém isat ion.69
HOOC NH
acide 0-glutamique
NaNo* HCI 2 N - H20 63%
La réaction procède avec rétention de configuration. En effet, le départ de l'ion
diazonium formé est assisté par l'hydroxyle du groupement COOH vicinal, conduisant à la formation d'un époxyde intermédiaire. L'ouverture de celui-ci
se fait par le deuxième COOH de la moléculet qui résulte en la formation de la y-butyrolactone, ayant conservé la configuration du carbone asymétrique

présent dans le synthon de départ. Les données pour le composé 46 sont en accord avec celles retrouvées dans la littérature?
Les hydrures métalliques sont sans aucun doute les réactifs les plus utiles
pour la réduction d'un acide carboxylique en alcool primaire.71 Le BzHs est un excellent réactif pour la réduction rapide et efficace d'acides carboxyliques aliphatiques et aromatiques, conduisant à des intermédiaires borates qui sont ensuite hydrolysés. Le B2H6 est fréquemment utilisé en solution dans le THF, dans lequel il existe comme complexe avec ce solvant. Le complexe borane- diméthylsulfure, également en solution dans du THF, est une alternative plus soluble et plus stable pour effectuer cette réduction, mais moins réactive.
Ainsi, la réduction du groupement acide se fait a I'aide du complexe borane-diméthylsulfure.72 Brown et collaborateurs73 ont démontré que les acides carboxyliques sont réduits plus rapidement par ce réactif que les y-
butyrolactones.
Bien que cette hydroxy-lactone soit disponible commercialement, son prix est
excessivement élevé. Par conséquent, il est plus avantageux de la produire au laboratoire. Les réactions permettant de la préparer s'opèrent facilement sur grande échelle. Les caractéristiques du composé 47 sont en accord avec celles retrouvées dans la littérature.70 On note en IR la disparition de la bande large caractéristique d'ur! groupement COOH et le remplacement de celle-ci par une bande plus intense et moins large indiquant la présence d'un OH. Nous avons maintenant en main le squeiette simplifié de la molécule à partir duquel nous viendrons greffer les fonctions manquantes.
L'hydroxy-lactone 47 est ensuite protégée à I'aide d'un groupement trialkylsilyle. Depuis l'introduction de ces groupements, la protection a I'aide de silyles est devenue de plus en plus répandue? Les sélectivités lors de la mise

en place et les diverses stabilités face au clivage ont résulté en un véritable essor d'applications. De nos jours, pratiquement toutes les synthèses totales comportent l'utilisation d'un groupement protecteur silylé a une étape ou l'autre
de la séquence.
Le choix du groupement protecteur s'avère à ce stadeci très important. Pour l'étape qui suivra, on cherche à générer un encombrement sur l'une des faces de la molécule afin d'obtenir une meilleure sélectivité. II s'agit donc de protéger le OH avec le groupement protecteur le plus volumineux possible.
Des essais avec le Ph3C se sont avérés infructueux puisque ce dernier était trop labile lors des étapes de purification (chromatographies sur gel de silice ou Florisil) des composés. L'utilisation du t-butyldiphénylsilyle (TBDPS)75.76 se revèle donc le choix le plus pratique pour cette synthese dû à sa stabilité et au grand encombrement qu'il génère. II s'installe et s'enlève facilement et résiste à de nombreuses conditions de réaction. On note en IR la perte de la bande
OH et l'apparition, en RMN 1H, des pics caractéristiques du TBDPS, soit les 10
hydrogènes aromatiques (multiplets à 7.36 et 7.67 ppm) et le t-butyle (singulet à 1.06 ppm). Les données pour le composé 48 sont en accord avec celles retrouvées dans la littérature.77
Ayant maintenant créé un encombrement sur le substituant en C-5, on prévoit que l'attaque d'un agent nucléophile se fera préférentiellement par la face la moins encombrée de la molécule, soit ici la face a. Beaucoup de
composés naturels d'importance biologique et d'intérêt synthétique possèdent un squelette hautement oxygéné. Les stratégies de synthese impliquant la construction d'un squelette carboné sous une forme simplifiée, puis l'installation subséquente des autres fonctions comportent certains attraits. En
ce sens, les travaux portant sur l'insertion de groupements hydroxyles,
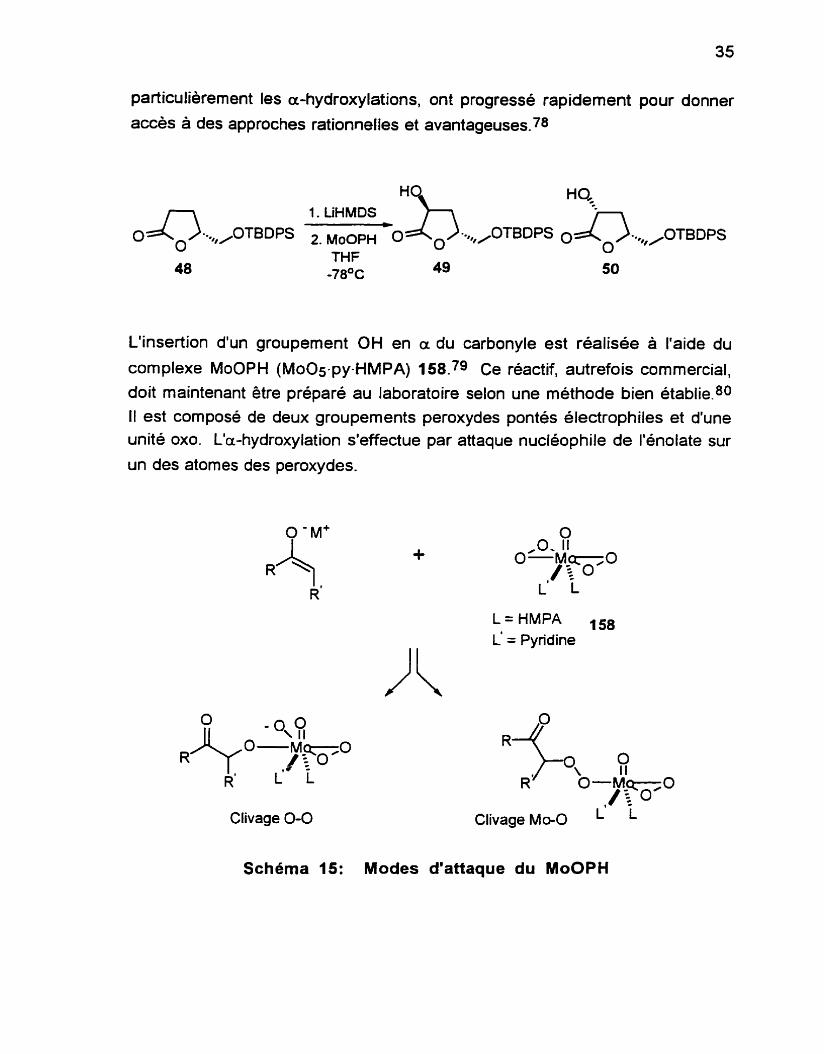
particulièrement les a-hydroxylations, ont progressé rapidement pour donner
accès à des approches rationnelles et avantageuses?
fi 0% 1. LiHMDS
- OTBDPS O O ,,,, n%,,OTBDPS 2 MaOPH O O
a- OTBDPÇ THF
48 - 7 8 ' ~ 49 50
L'insertion d'un groupement OH en a du carbonyle est réalisée à l'aide du
complexe MoOPH (MOOS-py-HMPA) 158.79 Ce réactif, autrefois commercial, doit maintenant être préparé au laboratoire selon une méthode bien établie? II est composé de deux groupements peroxydes pontés électrophiles et d'une unité 0x0. L'a-hydroxylation s'effectue par attaque nucléophile de I'énolate sur
un des atomes des peroxydes.
Clivage 0-0
L=HMPA 158 L' = Pyridine
Clivage Mo-O L- L
Schéma 15: Modes d'attaque du MoOPH

Deux modes d'attaque sont possibles. Cependant, comme des a-hydro-
peroxydes n'ont jamais été isolés, on suppose que le cheminement implique uniquement un clivage 0-0 (Schéma 15).
Le choix de la base s'avère important puisque des essais avec LDA et
Nai-iMDS ont conduit a des rendements faibles. L'utilisation de LiHMDS résulte en un rapport de 7:1 (déterminé par RMN 'H) en faveur de l'alcool 49
(64%).76 Les deux isomeres sont séparables par chromatographie sur gel de silice. L'alcool 49 est un solide blanc tandis que l'autre diastéréoisomère est une huile. Sur RMN 'H, on peut distinguer les deux isomeres: le H sur le C-3 est à 4.81 ppm pour l'alcool 49 tandis qu'on le retrouve à 4.53 ppm pour
l'isomère 50.
La sélectivité de cette réaction peut être expliquée selon le Schéma 16. L'encombrement crée par le TBDPS sur la face P de la molécule force le réactif à entrer préférentiellement par la face a.
(M~~N)~P=O.?+~P c M s O
Py- / 0-0
Schéma 16: Origine de la sélectivité avec le MoOPH
Des essais avec le (+)-tram-2-(p hénylsu1phonyl)-3-phényloxaziridine 159, développé par Davis,81 ont conduit à une sélectivité de 3.5:1, avec des rendements similaires à ceux obtenus avec le MoOPH, en utilisant le LiHMDS. L'emploi de NaHMDS ou de KHMDS n'a pas permis d'améliorer ces résultats.82 L'oxaziridine 1 59 se prépare aussi selon une méthode bien
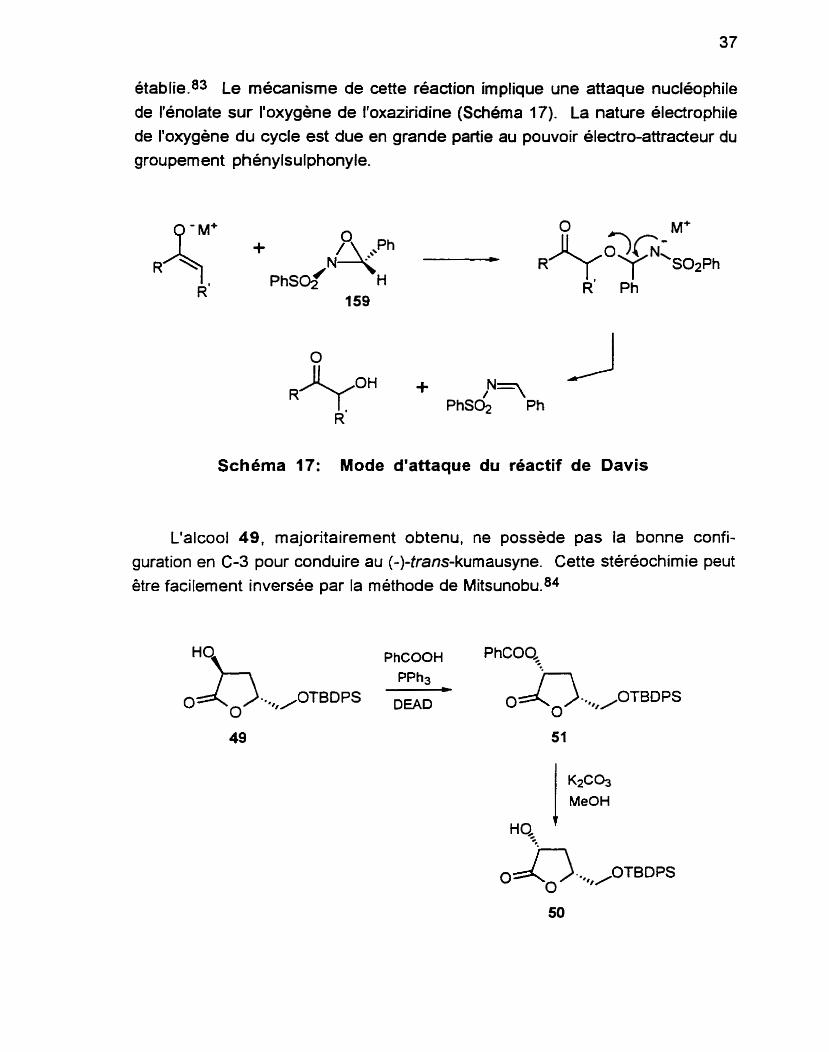
établie.83 Le mécanisme de cette réaction implique une attaque nucléophile de I'énolate sur I'oxygene de I'oxaziridine (Schéma 1 7). La nature électrophile de l'oxygène du cycle est due en grande partie au pouvoir électro-attracteur du groupement phénylsulphonyle.
Schéma 17: Mode d'attaque du réactif de Davis
L'alcool 49, majoritairement obtenu, ne possède pas la bonne confi- guration en C-3 pour conduire au (-)-trans-kumausyne. Cette stéréochimie peut être facilement inversée par la méthode de Mitsunobu.84
1 MeOH
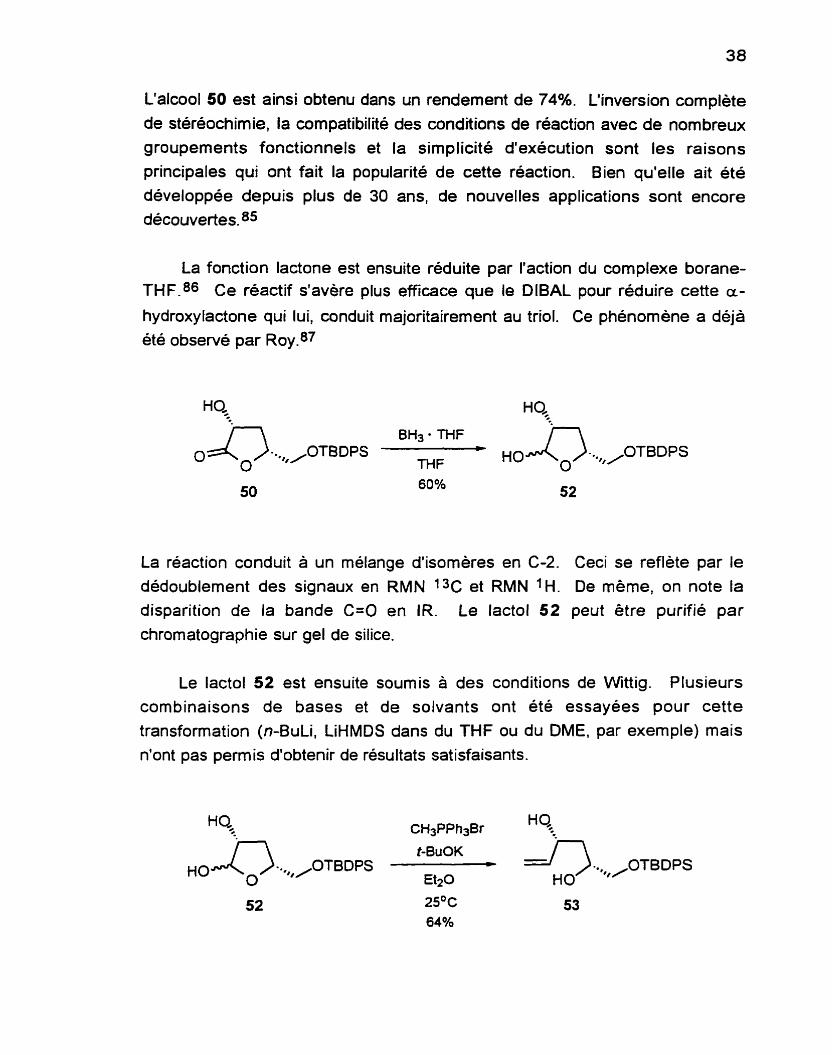
L'alcool 50 est ainsi obtenu dans un rendement de 7 4 % ~ L'inversion complète de stéréochimie, la compatibillé des wndlions de réaction avec de nombreux groupements fonctionnels et la simplicité d'exécution sont les raisons
principales qui ont fait la popularité de cette réaction. Bien qu'elle ait été
développée depuis plus de 30 ans, de nouvelles applications sont encore découvertes. 85
La fonction lactone est ensuite réduite par l'action du complexe borane- THF.86 Ce réactif s'avère plus efficace que le DIBAL pour réduire cette a-
hydroxylactone qui lui, conduit majoritairement au triol. Ce phénomène a déjà été observé par Roy.87
BH3 THF t
THF A-)- OTBDPS
Ho O **9/
La réaction conduit à un mélange d'isomères en C-2. Ceci se reflète par le dédoublement des signaux en RMN 1% et RMN 'H. De même, on note la disparition de la bande C=O en IR. Le lactol 52 peut être purifié par
chromatographie sur gel de silice.
Le lactol 52 est ensuite soumis à des conditions de Wittig. Plusieurs combinaisons de bases et de solvants ont été essayées pour cette
transformation (n-BuLi, LiHMDS dans du THF ou du DME, par exemple) mais n'ont pas permis d'obtenir de résultats satisfaisants.
A-)- OTBDPS t-BuOK
Ho O **+/ Et20 - =q- OTBDPS
HO '#, 0'

Une étude de Fiijer et de Quabeck88 a démontré l'efficacité du lBuOK dans les cas d'ylures non stabilisés. Leurs travaux portaient sur des méthylénations de cétones encombrées. Inspirés par les résultats de leurs travaux, nous avons choisi d'utiliser du PBuOK dans de l'éther. ce qui permet d'obtenir le 1.3diol anti 5 3 dans un rendement de 64%. comparable a ce qu'on peut retrouver dans la littérature89 pour des cas semblables. En RMN 1H. on note les pics caractéristiques de la double liaison terminale. soit le triplet dédoublé à 5.90 ppm et les deux doublets à 5.12 et 5.28 ppm.
Cette séquence comporte 6 étapes à partir du synthon commercial 47 et procède avec un rendement global de 15.5%. Afin d'éviter d'avoir à corriger en cours de route la stéréochimie du compose obtenu (i.e. alcool 4 9), nous avons développé, en parallèle à cette voie-ci, une deuxième méthode de préparation du diol 5 3, qui s'avère beaucoup plus efficace.
5. Deuxième voie de ~ ré~ara t ion du 1.3-diol anti
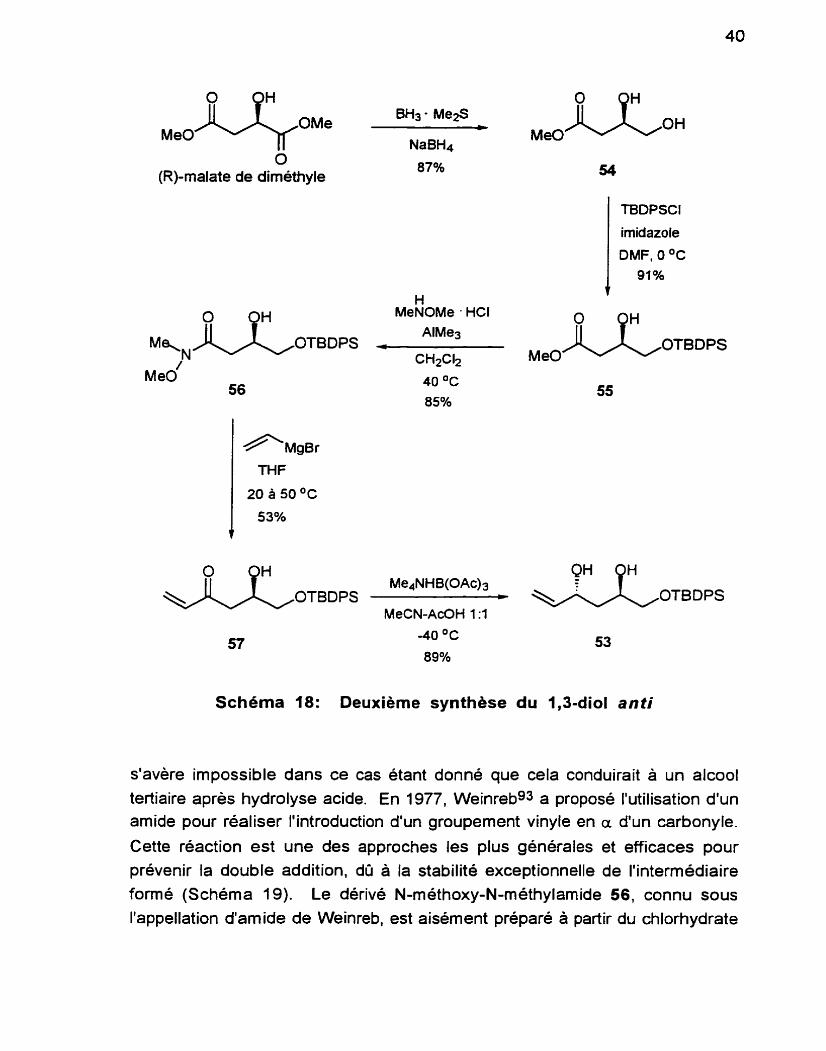
Cette séquence a été mise au point par loan-losif Radu (Schéma 18) et les détails expérimentaux de chaque étape sont contenus dans son mémoire de maîtrise.90 Ainsi. les réactions ne seront discutées que brièvement ici.
La première étape de cette séquence consiste en la réduction régioselective du (R)-malate de diméthyle, selon la méthode de Moriwake.91 Le composé 5 4 est obtenu, l'autre régioisomère n'ayant pas été détecté. En présence du complexe borane-diméthylsulfure, la réduction du groupement ester est très lente et incomplète. Par contre, si la réaction est effectuée avec le NaBH4 uniquement. la vitesse de la réaction augmente mais en défaveur de la régiosélectivité. Ainsi, l'addition catalytique de NaBH4 permet d'accélérer la réaction tout en conservant une sélectivité remarquable.
i a fonction hydroxyle primaire est ensuite protégée sélectivement à l'aide du groupement TBDPS.75.92 Pour accéder au diol 53. il est nécessaire a ce çtade-ci que la chaîne soit allongée de deux carbones, ou plus préciskment que le groupement vinyle soit mis en place. L'utilisation d'un réactif de Grignard
O (R)-malate de diméthyle
87%
1 imidazole
1 DMF, O OC
91%
M ~ N O M ~ - HCI
AIMe3
M% 1 CH2CI2 Me0
PM~B~ THF
Schéma 18: Deuxième synthése du 1,3-diol anti
s'avère impossible dans ce cas étant donné que cela conduirait à un alcool
tertiaire après hydrolyse acide. En 1977, Weinrebgs a proposé l'utilisation d'un amide pour réaliser l'introduction d'un groupement vinyle en a d'un carbonyle.
Cette réaction est une des approches les plus générales et efficaces pour prévenir la double addition, dû à la stabilité exceptionnelle de l'intermédiaire
formé (Schéma 19). Le dérivé N-méthoxy-N-méthylarnide 56, connu sous l'appellation d'amide de Weinreb, est aisément préparé à partir du chlorhydrate

de N,O-dirnéthylhydroxylarnine commercial et du composé 55. Ainsi, même si un excès d'organomagnésien ou lithien est utilise, l'alcool tertiaire ne se forme
pas et les conditions réactionnelles pour cette transformation sont considérées
comme très douces. L'amide de Weinreb 56, ainsi mis en présence d'un excès de bromure de vinylmagnésien conduit a la vinyl-cétone 57, dans un
rendement acceptable de 53%.
II R'M R-C, ,O-CH3 -
N I THF
CH3 amide de Weinreb
Schéma 19: Amide de Weinreb
La fonction carbonyle est ensuite réduite sélectivement avec le
Me,+NHB(OAc)3, selon les conditions de Evan~.~48gs Le diol anfi 53 est obtenu
dans un rendement de 89% et aucune trace de son isomère n'a pu être
détectée. Le mécanisme proposé pour cette réaction fait intervenir un
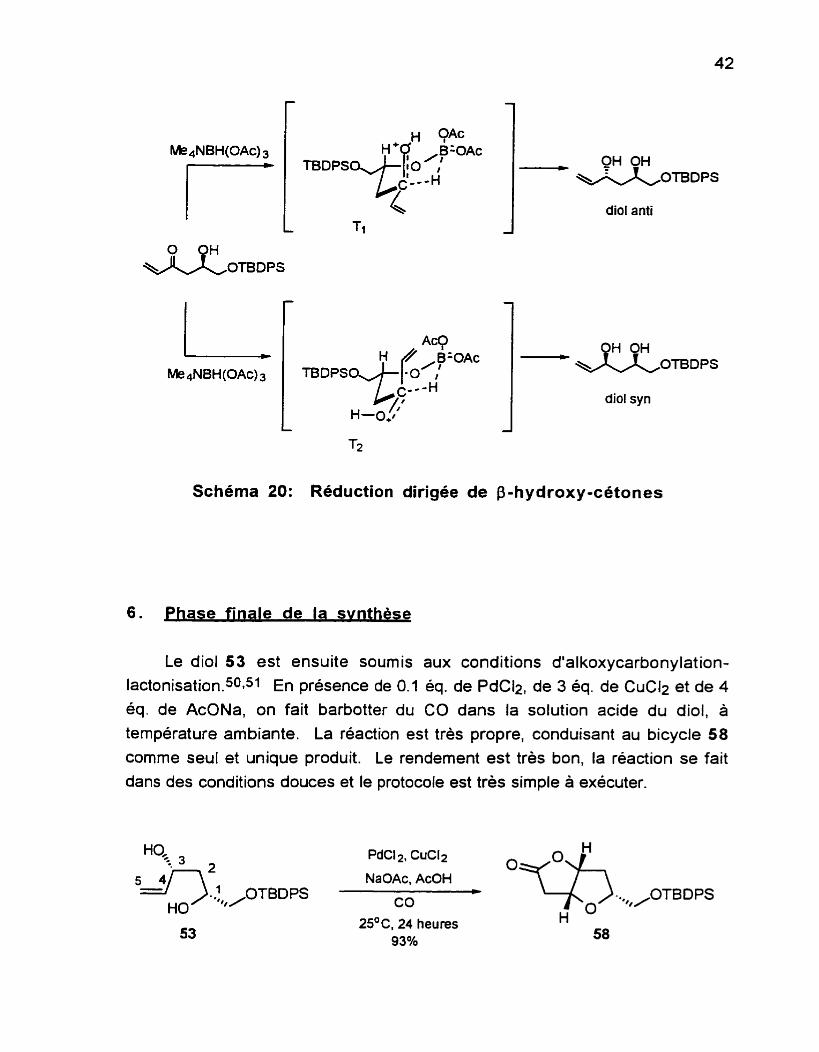
intermédiaire cyclique à 6 membres (Schéma 20), formé par l'échange d'un
ligand acétate sur l'anion triacétoxyborohydrure par la fonction hydroxyle du substrat (catalysée par un proton du solvant). Des deux états de transition
possibles, le T l est favorise à cause de l'oxygène du carbonyle en position axiale (effets stéréoélectroniques favorables). Également, le T2 est défavorisé dû à l'interaction diaxiale-1,3 entre le groupement vinyle et le groupement
acétyle. Cette interaction est plus importante ici que celle entre le H du C=O et l'acétyle dans le Tl. II s'ensuit une livraison intramoléculaire d'un hydrure à partir du bore vers le carbonyle pour former le diol anti exclusivement. Nous
verrons au prochain chapitre qu'il est possible d'obtenir, à partir du composé 57, le diol syn en utilisant d'autres conditions réa~tionnelIes.95~96
Cette nouvelle voie menant au di01 53 procède donc en 5 étapes avec un rendement global de 31.7% à partir du (R)-rnalate de diméthyle.
diol anti
-= OTBDPS
diol syn
Schéma 20: Reduction dirigée de 8-hydroxy-cétones
6 . Phase finale de la svnthèse
Le diol 53 est ensuite soumis aux conditions d'alkoxycarbonylation- lactonisation.50*51 En présence de 0.1 éq. de PdC12, de 3 éq. de CuC12 et de 4
éq. de AcONa, on fait barbotter du CO dans la solution acide du diol, à température ambiante. La réaction est tres propre, conduisant au bicycle 58 comme seul et unique produit. Le rendement est très bon, la réaction se fait dans des conditions douces et le protocole est tres simple à exécuter.
HO, 5 3 PdCl 2, CU CI^
5 4 n 2 . NaOAc, AcOH d )-.~,,,OTBDPS -
HO CO y,/ -*.,f /OTBDPS
L- 25%, 24 heures H
ce
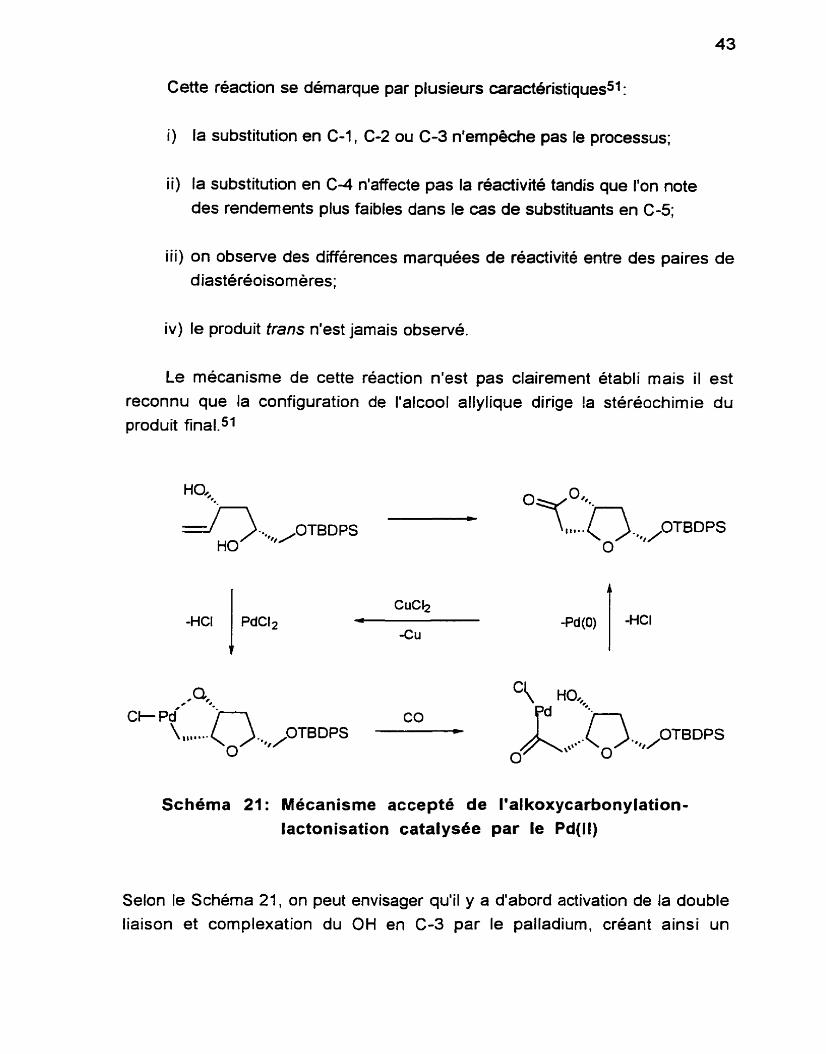
Cette réaction se démarque par plusieurs caracteristiques5?
i) la substitution en C-1, C-2 ou C-3 n'empêche pas le processus;
ii) la substitution en C-4 n'affecte pas la réactivité tandis que l'on note des rendements plus faibles dans le cas de substituants en C-5;
iii) on observe des différences marquées de réactivité entre des paires de
diasteréoisorneres;
iv) le produit tram n'est jamais observé.
Le mécanisme de cette réaction n'est pas clairement établi mais il est reconnu que la configuration de l'alcool allylique dirige la stéréochimie du produit final.51
-Pd (O)
HO,,
Schéma 21 : Mécanisme accepté de I'alkoxycarbonylation- lactonisation catalysée par le Pd(ll)
Selon le Schéma 21, on peut envisager qu'il y a d'abord activation de la double liaison et complexation du OH en C-3 par le palladium, créant ainsi un

encombrement sur l'une des faces de la double liaison. Cette complexation dirige la formation d'une jonction de cycle cis uniquement. Le groupement hydroxyle en C-1 ne peut donc attaquer la double liaison nouvellement activée
que d'un seul côté, pour conduire ainsi à la formation de la portion furane du bicycle. II y a ensuite insertion de monoxyde de carbone puis fermeture du
deuxième cycle.
En IR, on note l'apparition de la bande C=O (1780 cm-'). En RMN IH, on distingue aisément les deux hydrogènes sur la jonction de cycle, soient les systèmes à 5.00 et 4.57 ppm.
Les étapes qui suivent permettent de mettre en place la chaîne trans-ene- yne.
La portion lactone est réduite avec DIBAL. La réaction procède aisément
et très rapidement, avec un rendement quantitatif. Dans ce cas également, on
obtient un mélange de diastéréoisomères. Le composé peut être purifié par chromatographie sur gel de silice.
.*#,,' OTBOPS THF
-78OC .'9/ OTBDPS
Le lactol 59 est ensuite traité avec un excès de I'ylure dérivé du bromure de (3-triméthylsilyl-2-propyny1)-triphénylposponi disponible commer- cialement, pour conduire au composé 60 dans un rendement de 85%, ainsi qu'une petite quantité de l'isomère ayant la double liaison de géométrie cis. Ces deux composés sont séparables par chromatographie sur gel de silice. Sur RMN 1 H, on distingue les deux composés a l'aide de la géométrie de la double liaison. Dans l'isomère trans, le système à 5.66 ppm est un doublet avec une constante de couplage de 16.0 Hz tandis que dans l'isomère cis, le système correspondant se situe a 5.63 ppm avec une constante de couplage
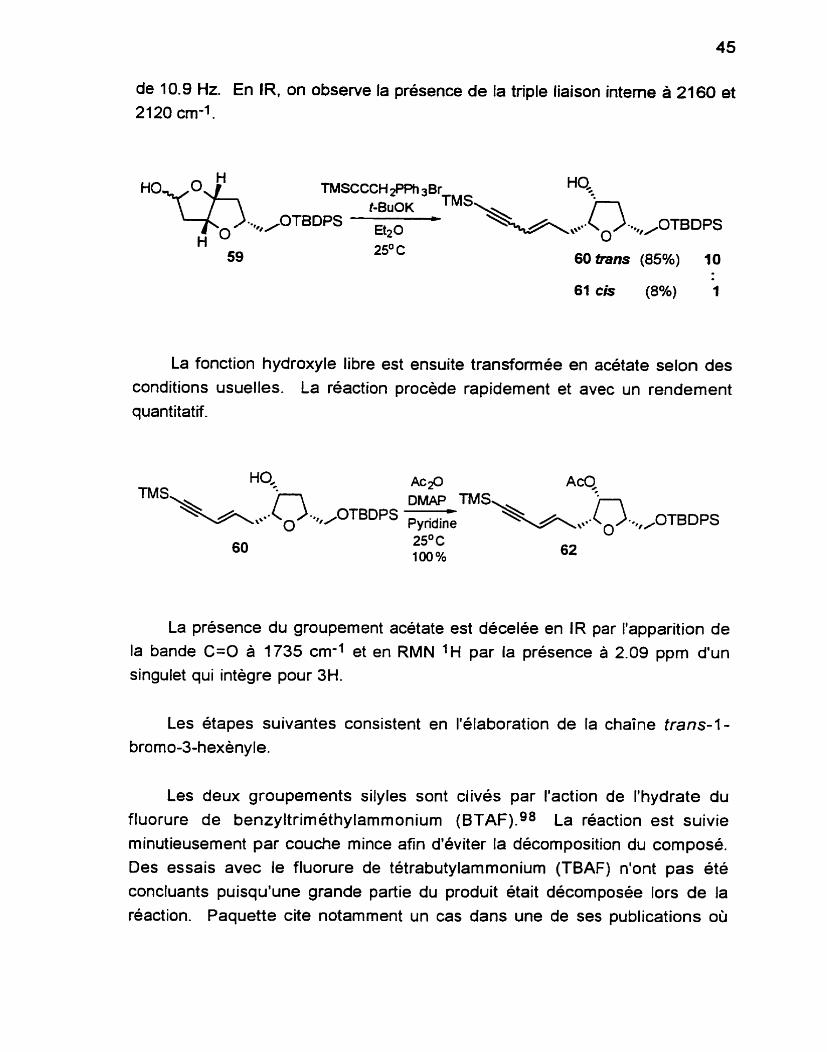
de 10.9 Hz En IR, on observe la présence de la triple liaison interne a 2160 et
2120 cm-'.
TMSCCCH 2 P ~ HO, t-BuOK
OTBDPS UzO / 3's\,,,.b- 0 .+t / OTBDps
25' C 60 &ans (85%) 10
La fonction hydroxyle libre est ensuite transformée en acétate selon des conditions usuelles. La réaction procède rapidement et avec un rendement quantitatif.
La présence du groupement acétate est décelée en IR par l'apparition de la bande C=O à 1735 cm-' et en RMN 1H par la présence a 2.09 ppm d'un singulet qui intègre pour 3H.
Les étapes suivantes consistent en l'élaboration de la chaîne trans-l- bromo-3-hexenyle.
Les deux groupements silyles sont clivés par l'action de l'hydrate du fluorure de benzyltriméthylammonium ( B T A F ) . ~ ~ La réaction est suivie minutieusement par couche mince afin d'éviter la décomposition du composé. Des essais avec le fluorure de tétrabutylammonium (TBAF) n'ont pas été concluants puisqu'une grande partie du produit était décomposée lors de la
réaction. Paquette cite notamment un cas dans une de ses publications ou
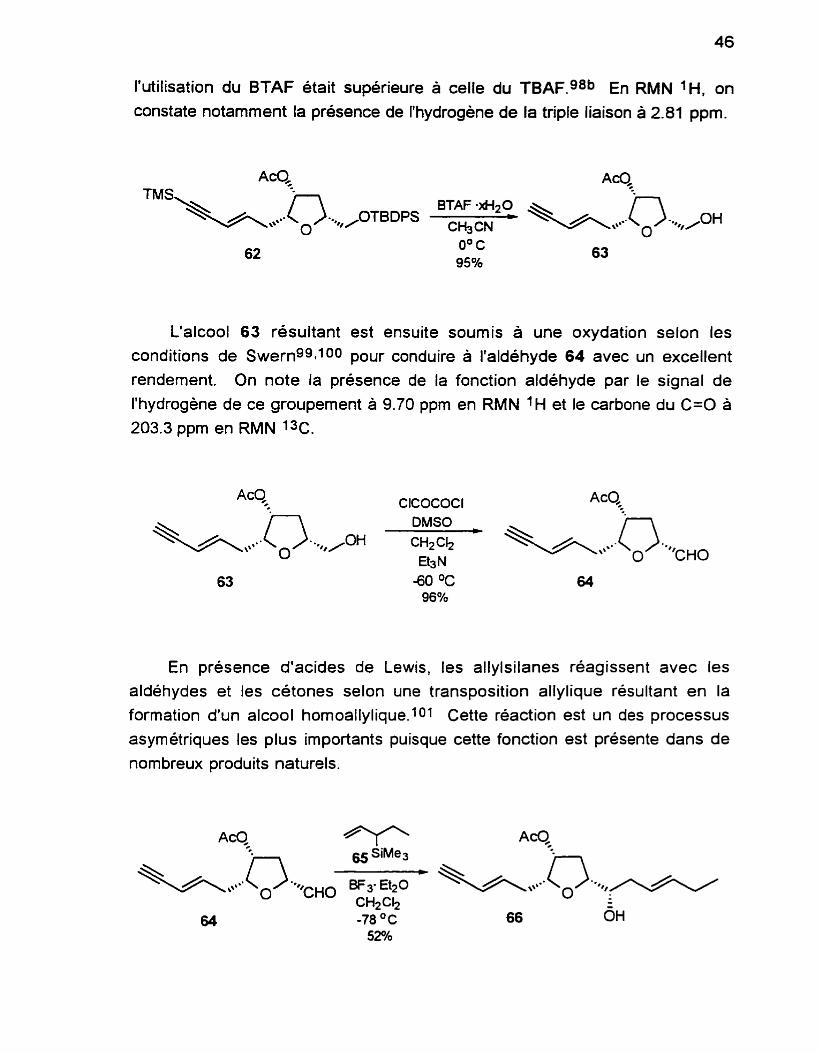
l'utilisation du BTAF était supérieure à celle du TBAF.g8b En RMN 'H, on constate notamment la présence de l'hydrogène de la triple liaison à 2.81 ppm.
L'alcool 63 résultant est ensuite soumis à une oxydation selon les
conditions de SwernggIloo pour conduire à l'aldéhyde 64 avec un excellent
rendement. On note la présence de la fonction aldéhyde par le signal de
l'hydrogène de ce groupement à 9.70 ppm en RMN ' H et le carbone du C=O à
203.3 ppm en RMN 1%.
En présence d'acides de Lewis, les allylsilanes réagissent avec les
aldéhydes et les cétones selon une transposition allylique résultant en la
formation d'un alcool homoallylique.1o1 Cette réaction est un des processus
asymétriques les plus importants puisque cette fonction est présente dans de
nombreux produits naturels.
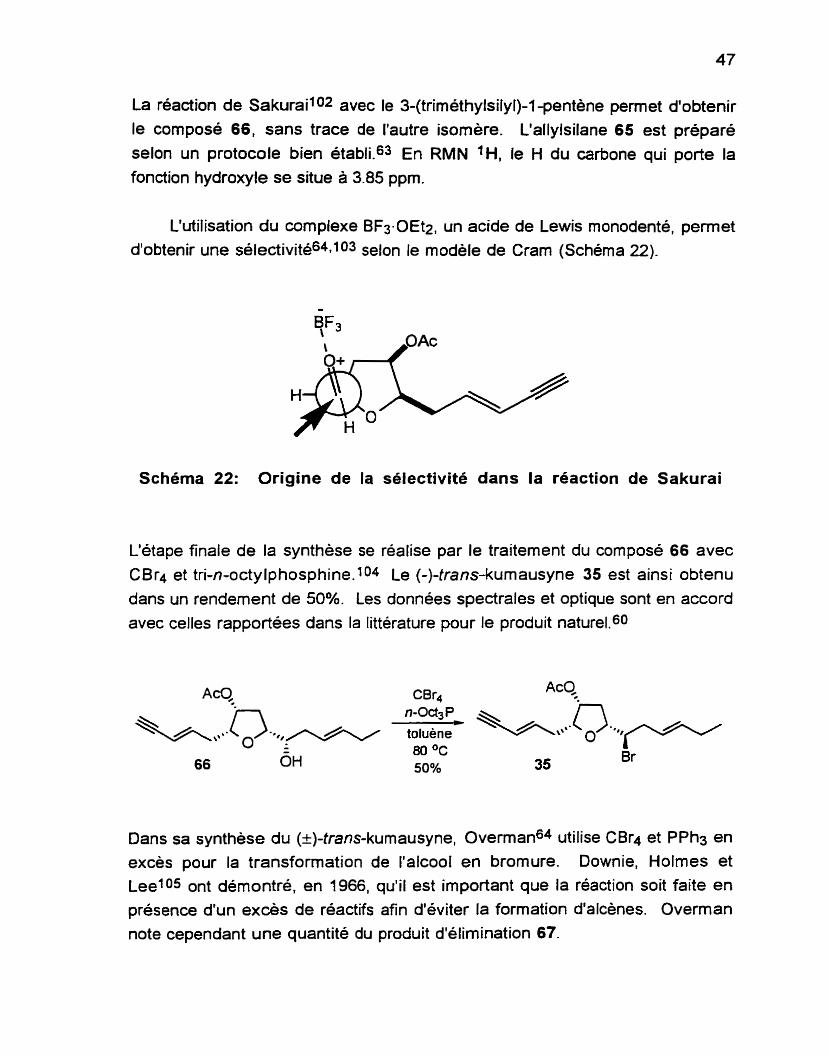
La réaction de Sakuraii02 avec le 3-(triméthylsily1)-l -pentène permet d'obtenir
le composé 66, sans trace de l'autre isomère. L'allylsilane 65 est préparé selon un protocole bien établi.63 En RMN 'H, le H du carbone qui porte la fonction hydroxyle se situe à 3.85 ppm.
L'utilisation du complexe BF3-OEt2, un acide de Lewis monodenté, permet d'obtenir une sélectivité6491o3 selon le modèle de Cram (Schéma 22).
Schéma 22: Origine de la sélectivité dans la réaction de Sakurai
L'étape finale de la synthèse se réalise par le traitement du composé 66 avec
CB r4 et tri-n-octylp hosp hine. 104 Le (-)-trans-kumausyne 35 est ainsi obtenu dans un rendement de
avec celles rapportées
50%. Les données spectrales et optique sont en accord dans la littérature pour le produit naturel.60
Am, CBr4 AcQ
*
o.. n-0d3 P \\\,*.
- toluène +K&.\,~. Q,,-
O "J 80 O C on 35 Br 66 50%
Dans sa synthèse du (+)-trans-kumausyne, 0vemanE4 utilise CBr4 et PPh3 en excès pour la transformation de l'alcool en bromure. Downie, Holmes et
Leelos ont démontré, en 1966, qu'il est important que la réaction soit faite en présence d'un excès de réactifs afin d'éviter la formation d'alcènes. Overman
note cependant une quantité du produit d'élimination 67.

À la lumière de ces résultats, nous avons choisi d'utiliser le n-0ctaP. Bien que la PPh3 soit moins basique que les trialkylphosphines, ces derniers sont de
meilleurs nucleophiles.~06 Malgré le fait que nous n'ayons pu déterminer si le produit d'élimination était présent ou non dans le brut de notre réaction, il n'en demeure pas moins que le bromure est obtenu dans un meilleur rendement
selon les conditions expérimentales que nous avons utilisées. Pour sa synthèse du (+)-laurencin 39 (voir Schéma 8), Murai107 a également utilisé ces conditions pour aboutir au bromure avec un très bon rendement.
Le présent cheminement menant au (-)-trans-kumausyne comporte 13
étapes a partir du (R)-malate de diméthyle avec un rendement global de 5.95%.
II est considérablement plus court et plus direct que les autres synthèses décrites à ce jour.108
À titre comparatif, citons l'exemple de Lee et collaborateursl*g qui ont
réalisé tout demièrement une synthèse formelle du (-)-k~mausallene,~~o basée sur la cyclisation radicaiaire de P-alkoxyacrylates (Schéma 23). L'étape-clé
conduit à un mélange de deux bicycles, avec un bon rendement.
Notre stratégie ne donne accès qu'a un seul et unique composé, avec un excellent contrôle de la stéréochimie et un excellent rendement. II serait possible également d'appliquer notre approche à la synthèse du (-)-
kurnausallène.
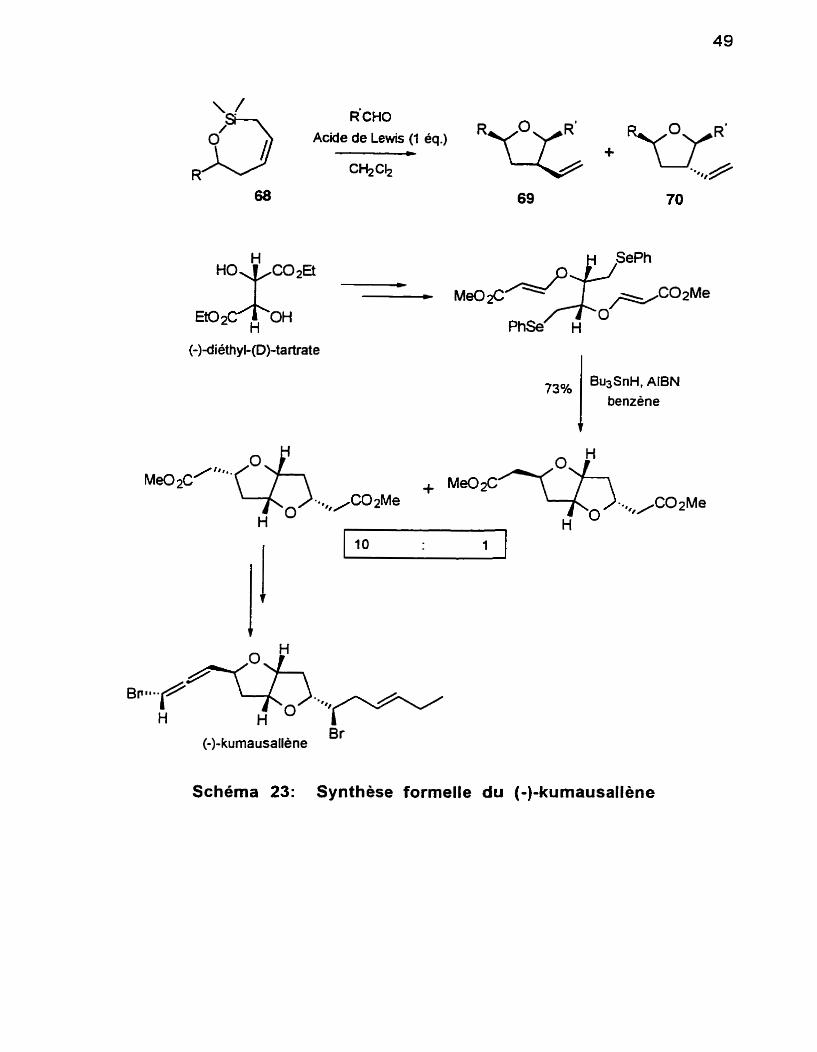
R'CHO Acide de Lewis (1 éq.)
t
R c w i ,
73% Bu3SnH, AIBN
benzène
Schéma 23: Synthèse formelle du (-)-kumausallène

CHAPITRE 2
Vers la synthèse totale d'un métabolite des algues brunes Notheia anomala
Tel que décrit au chapitre précédent, les nombreuses espèces d'algues rouges ont été beaucoup étudiées. A l'opposé, les algues brunes ont reçu beaucoup moins d'attention. Commercialement, elles sont récoltées pour leur contenu riche en polysaccharides.
La chimie des algues brunes est dominée par les métabolites secondaires, qui sont en majorité des terpènes, plus particulièrement des diterpènes.10112 Ces composés se distinguent par une grande variété de structures et sont souvent polyoxygénés. On y retrouve des époxy-lipides qui démontrent des similarités structurales avec des phéromones connues. On a ainsi supposé, par analogie, que ces composés sont des messagers chimiques
(régulateurs de croissance, par exemple) et de nombreuses études ont été faites pour établir le rôle de ces métabolites ainsi que leur mécanisme de fonctionnement.
En 1980, Warren et collaborateurs61 ont étudié les constituants des algues
brunes Notheia anornala, membres de la famille des Notheiaceae (ordre des Chondaliales). Ces algues, récoltées sur les côtes d'Australie (Torquay, Victoria), croissent de manière épiphyte sur les algues brunes Hormosira banksii (ordre Fucales). Le (6s. 7s. 9R, 1 0R)ô,9-6poxynonadèc-18-ene-7, I O -
diol 36 s'y retrouve en majorité (Schéma 24). La structure a été assignée sur la base de données spectrales et optiques et confirmée par rayons-X. Récemment, on a découvert que cette famille de tétrahydrofuranes étaient des inhibiteurs puissants et sélectifs du développement au stade larvaire de
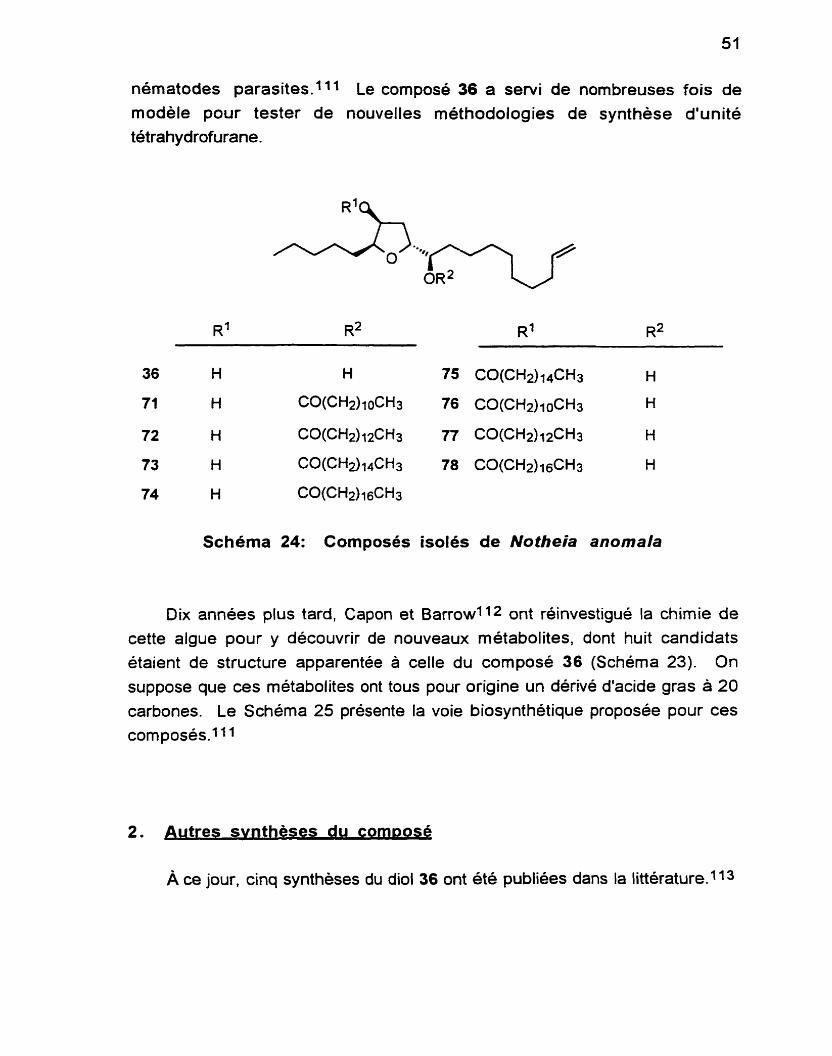
nématodes parasites.lll Le compose 36 a servi de nombreuses fois de
modèle pour tester de nouvelles méthodologies de synthèse d'unité tétrahydrofurane.
Schéma 24: Composés isolés de Notheia anomala
Dix années plus tard, Capon et Barrow1 12 ont réinvestigué la chimie de
cette algue pour y découvrir de nouveaux métabolites, dont huit candidats étaient de structure apparentée à celle du composé 36 (Schéma 23). On
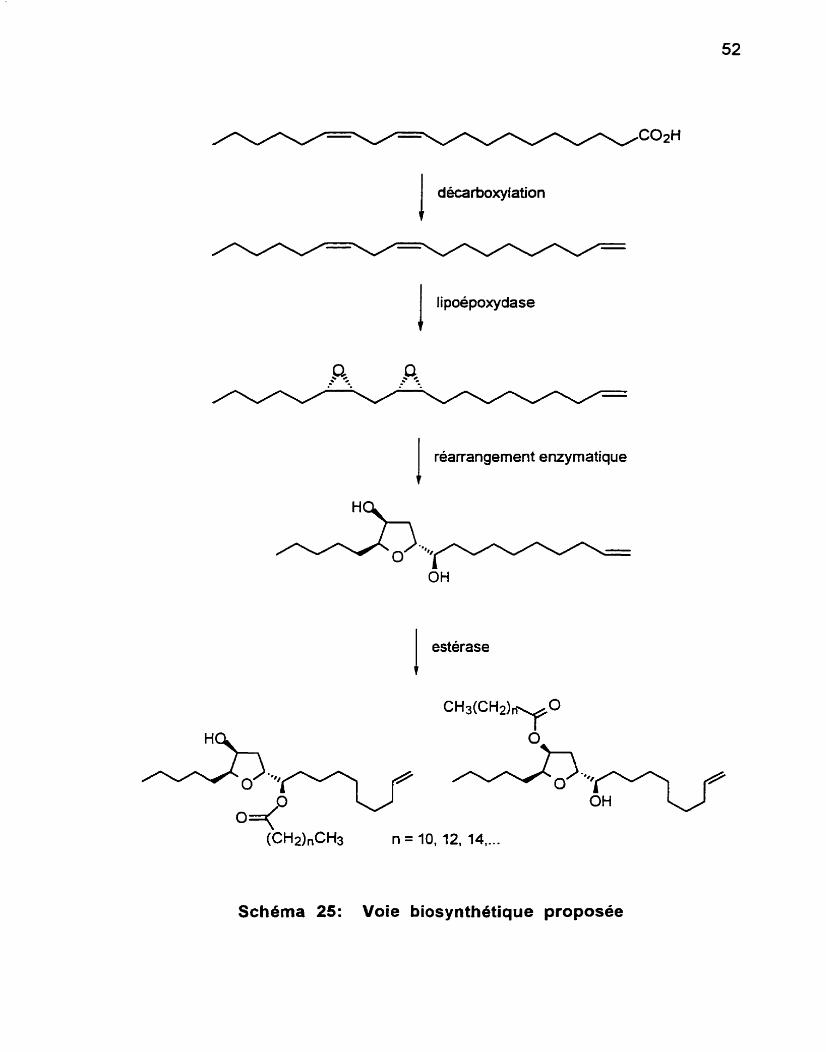
suppose que ces métabolites ont tous pour origine un dérivé d'acide gras à 20 carbones. Le Schéma 25 présente la voie biosynthétique proposée pour ces composés.1
tres svnthèses du com~osé 2. #lu
À ce jour, cinq synthèses du diol 36 ont été publiées dans la littérature.ll3
1 Iipoépoxydase
réarrangement enzymatique
1 estérase
O -foi /-& -oOI(v,y 0 4
OH
(CH2hCH3 n = IO, 12, 14, ...
Schhma 25: Voie biosynthétique proposée

En 1984, Williams et collaborateürs~oo ont préparé le (f)-diol 36 selon
une séquence faisant appel à l'oxydation par NBS d'un précurseur 1'3-dioxo-
lane, suivie d'un couplage de Grignard (Schéma 26). Cette voie procède en 14
étapes (bien que les auteurs n'en fassent pas mention. le produit de départ doit
provenir du couplage entre le l-heptyne et le dérivé benzylique du glycidol) et malgré que le rendement soit élevé pour plusieurs étapes, une résolution
chromatographique de stéréoisomères est requise afin d'obtenir le précurseur
1,3-dioxolane requis pour I'étapeclé. La réaction conduit en effet aux quatre diastéréoisomères qui sont séparés et soumis individuellement aux étapes
subséquentes. L'identification sans ambiguite du précurseur nécessaire à cette
synthese n'a été possible qu'une fois la synthèse du composé 36 complétée, et ce, par comparaison scrupuleuse des quatre tetrahydrofuranes obtenus. De
plus, I'étapeclé conduit à un m édiocre rendement du tétrahydro-furane désiré.
Cette première réalisation a été suivie, en 1985, par la synthese
énantiosélective de Takano et collaborateurs,~l qui avait comme point de
départ le L-tartrate (Schéma 27). La chaîne décèn-01 est également placée grâce à un couplage de Grignard. Dans ce cas également, on doit faire appel à une séparation chromatographique pour résoudre les mélanges de stéréoiso- mères. Cette séquence procède en 16 étapes (-17%).
Une seconde synthèse énantioséiective a été réalisée en 1990 par
Gurjar et collaborateurs1 5 (Schéma 28). Cette voie débute avec un dérivé du
D-glucose protégé et aboutit à un intermédiaire connu, soit l'aldéhyde obtenu dans la synthèse de Williams ou encore celui obtenu dans celle de Takano.
Ceci constitue donc une synthèse formelle du di01 36. L'utilisation du D- glucose implique que l'on doit déoxygéner en C-3 ce chiron de départ.
En 1993, Chikashita et collaborateur^^^^ ont élaboré une autre voie vers
le di01 36, qui implique une cyclodéshydratation d'un diol-1,4, débutant avec le (S)-glycidol (Schéma 29). La réduction finale du composé avec NaBH4 conduit a une sélectivité de 4.2:l pour l'alcool désiré. Ceci implique donc que les diastéréoisomères doivent être séparés. Cette séquence procède en 10 étapes
avec un rendement global de -3%.
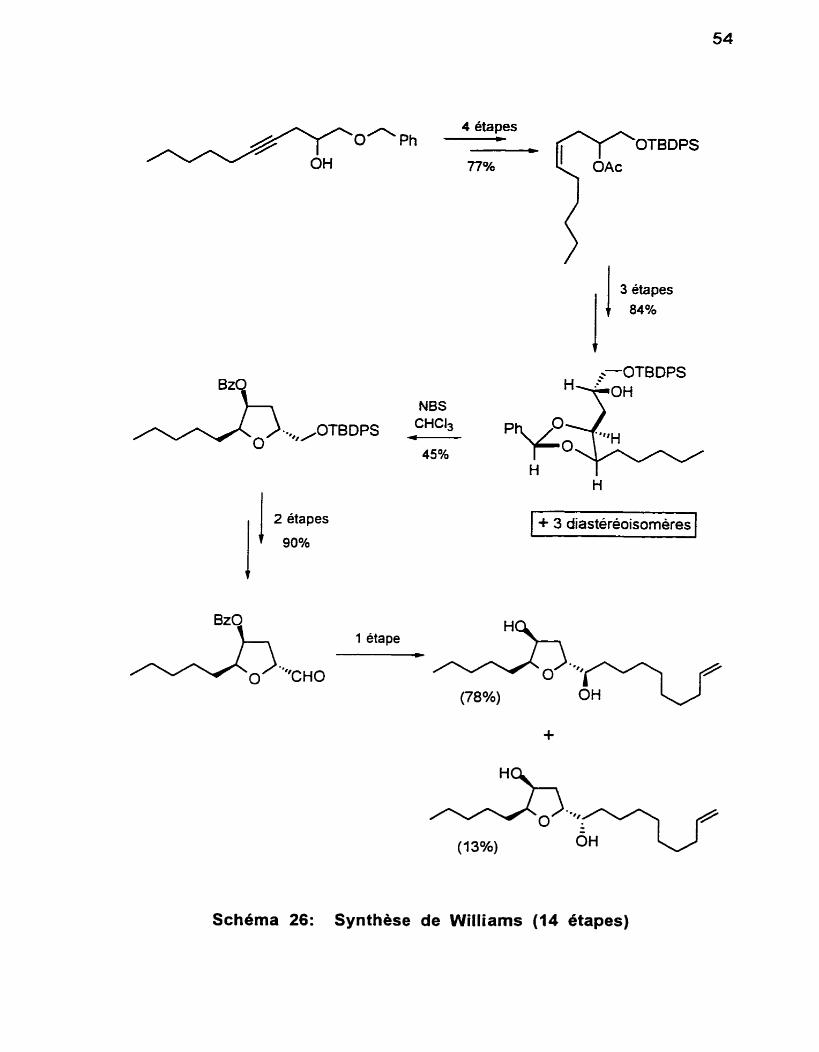
OH n./o OAC
2-OTBDPS
NBS CHCI3 ~ - * , , , , o T B D P s _ 45% pyIL
H
2 étapes
90% + 3 diastéréoisomères
Schhma 26: Synthése de Williams (14 étapes)
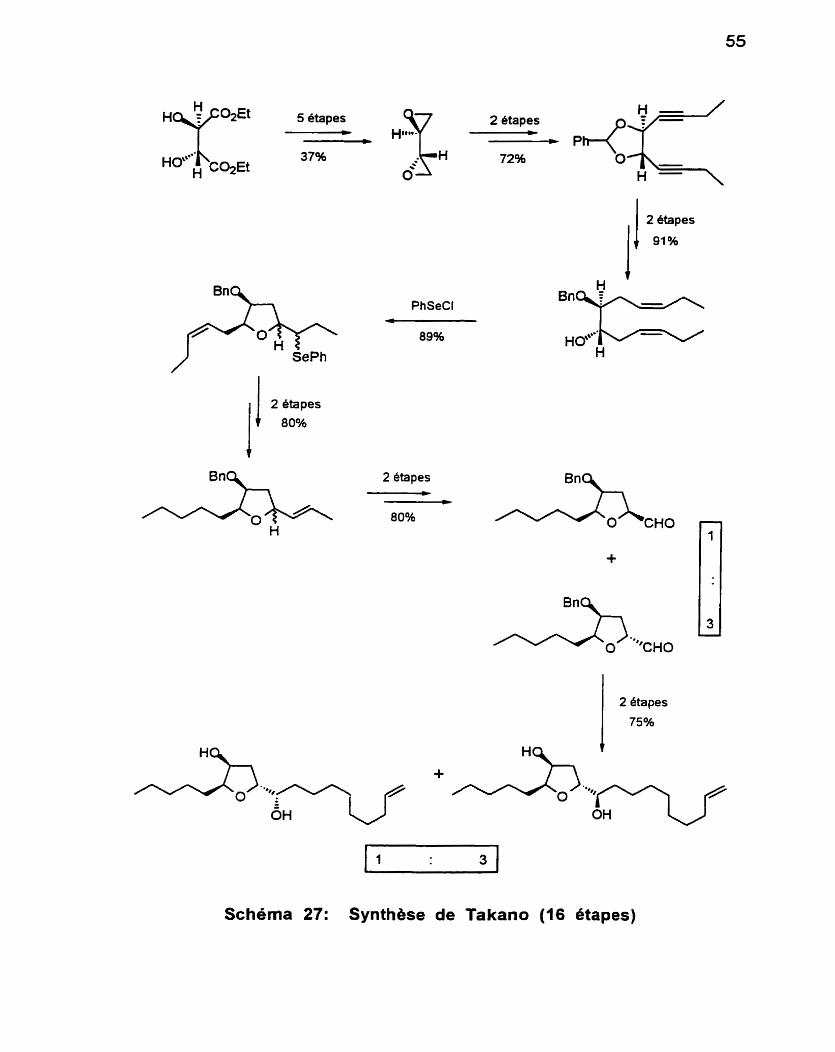
2 étapes
91 %
2 étapes
80°h
2 étapes - Y-
2 étapes
75 Or6
Schéma 27: Synthèse de Takano (16 étapes)
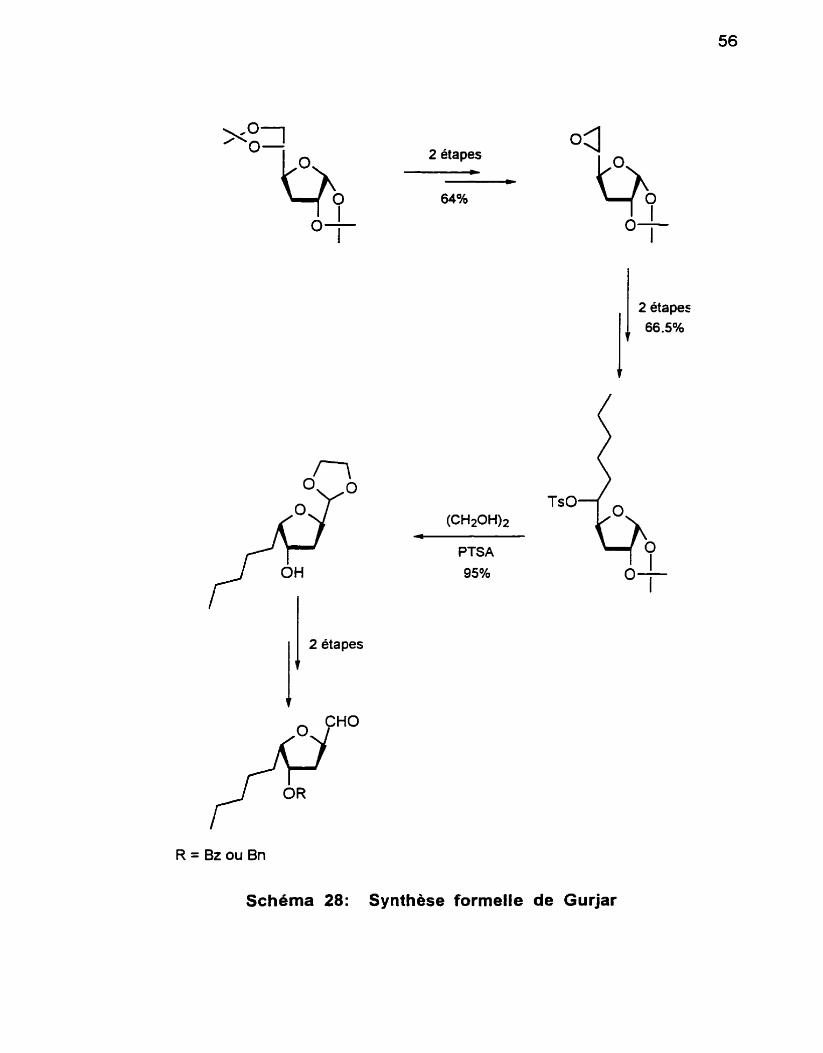
2 étapes - - 64Oh
s 2 étapes
2 étapes 66 .Sa!
Schéma 28: Synthèse formelle de Gurjar
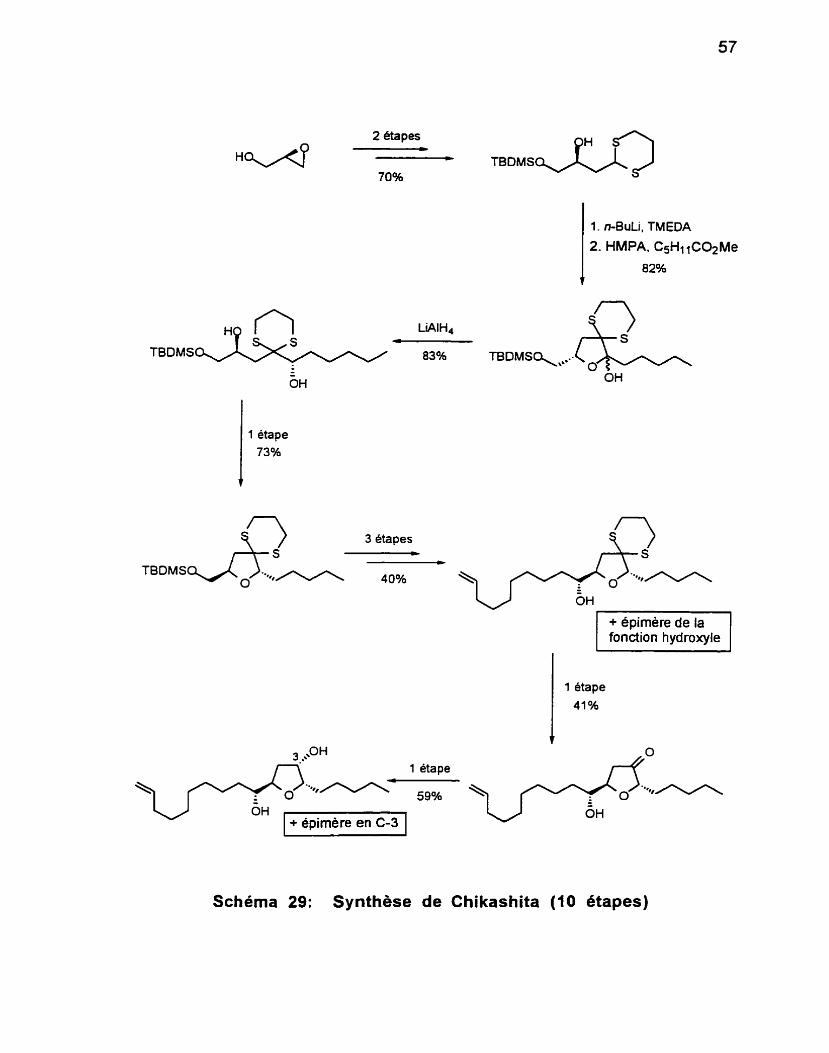
1. n-BuLi, TMEDA
2. HMPA, CSHIICQMe
LiAIH4
TBDMS 83% TBDMSq,,,..
OH OH
1 étape 73%
1
3 étapes
TBDMS 40%
I 1 étape
41 O h
,OH 1 étape
59% 4 OH 1 + épimère en C-3 1 &l
I I
Schéma 29: Synthèse
u OH
de Chikashita (10 étapes)

Tout récemment, Capon et Barrow117 ont présenté une approche biomimétique a la préparation des métabolites de Notheia anomala. Celle-ci repose sur une conversion de bis-époxydes en milieu acide, conduisant ainsi à
des tétrahydrofuranes. Le système modèle choisi pour cette étude est
représenté au Schéma 30. Les deux bis-époxydes cis-cis obtenus sont séparables par HPLC. Le traitement individuel de ces derniers en milieu acide acétique glacial conduit a la formation des tétrahydrofuranes (à noter que ces composes sont racémiques).
2 étapes v -OH -
1 2 étapes
. - 1 étape
l 1 2 étapes
Schema 30: Approche biomimétique de Capon et Barrow
Le mécanisme proposé est présenté au Schéma 31. La stéréochimie
relative des composés obtenus a été déterminée par comparaisons spectroscopiques avec le produit naturel 36.
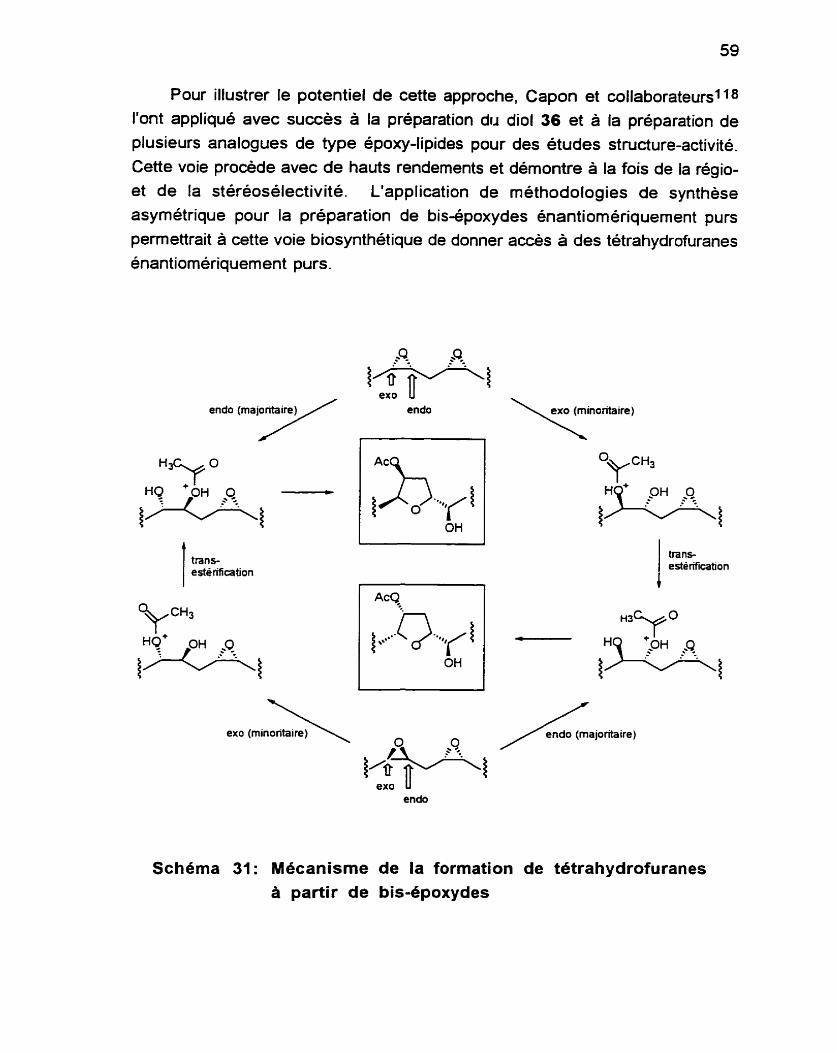
Pour illustrer le potentiel de cette approche, Capon et wllaborateursl l8
l'ont appliqué avec succès à la préparation du diol 36 et à la préparation de plusieurs analogues de type époxy-lipides pour des études structure-activité. Cette voie procède avec de hauts rendements et démontre à la fois de la régio- et de la stéréosélectivité. L'application de méthodologies de synthèse asymétrique pour la préparation de bisépoxydes énantiomériquernent purs
permettrait à cette voie biosynthétique de donner accès à des tétrahydrofuranes énantiomériquement purs.
endo (majontaire) /
1 trans- estérification
exo U endo exo (minontaire)
t , \
exo (minoritaire) \ endo (majontaire)
exo endo
Schéma 31 : Mecanisme de la formation de tetrahydrofuranes a partir de bis-époxydes
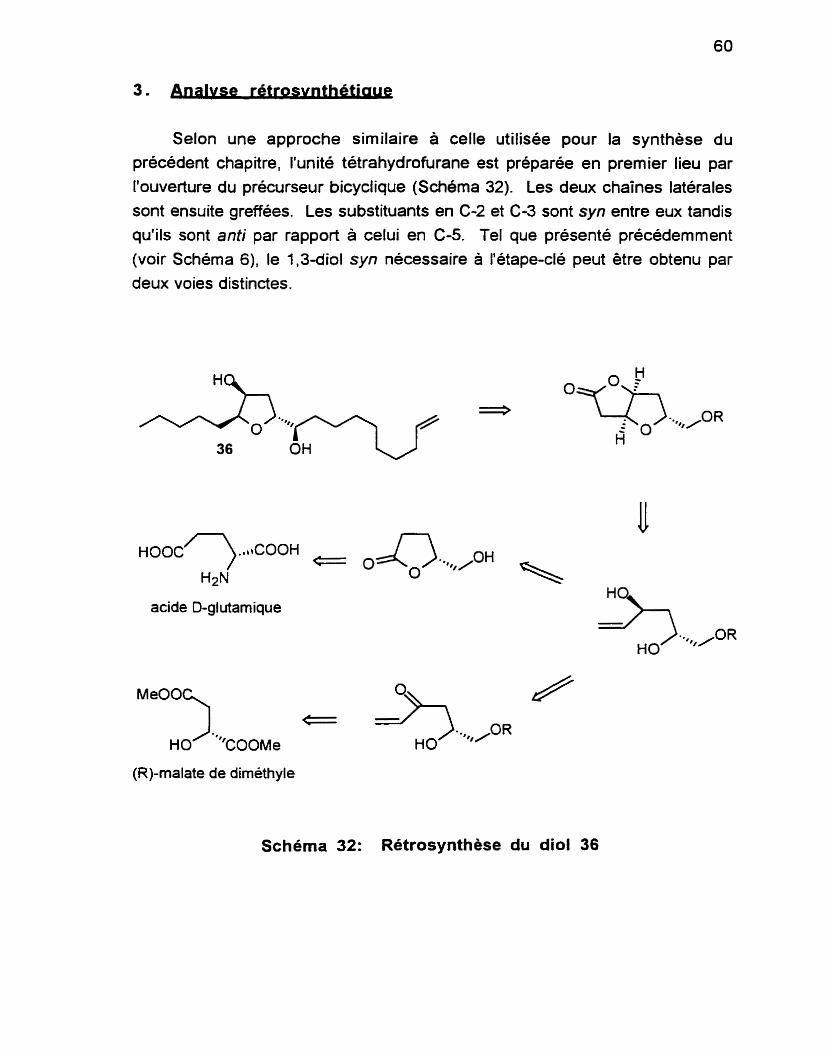
Selon une approche similaire à celle utilisée pour la synthèse du précédent chapitre, l'unité tétrahydrofurane est préparée en premier lieu par l'ouverture du précurseur bicyclique (Schéma 32). Les deux chaines latérales sont ensuite greffées. Les substituants en C-2 et C-3 sont syn entre eux tandis qu'ils sont anti par rapport à celui en C-5. Tel que présenté précédemment (voir Schéma 6), le 1,3-diol syn nécessaire à l'étape-clé peut être obtenu par
deux voies distinctes.
H2N
acide D-glutamique
e " ~ ~ " ' / c o o M e c==
(R)-malate de diméthyle
Schéma 32: Rétrosynthèse du di01 36

4. Premihre voie de synfbese du 1.3-diol svn
Le diol 80 peut être obtenu à partir du (R)-malate de diméthyle selon une
voie similaire à celle utilisée au chapitre précédent (voir P. 40). Dans ce cas, le composé 57 est traité avec le diéthylméthoxyborane conjointement avec le
NaB H4 pour obtenir cette fois, le 1,3diol syn 80 uniquement.gs La présence de l'autre isomère n'a pas été détectée.
La sélectivité96 de cette réaction s'explique par le fait qu'il y a complexation avec le Et2BOMe et l'hydrure est livré de manière externe par le
NaBH4 sur la face Re (Schéma 33).
THF / MeOH
92%
Le composé 80 et son isomère 53 sont facilement différenciés en RMN
'H. En effet, pour le diol syn 80, le signal du proton en C-3 est à 4.43 pprn
tandis qu'il se situe à 4.37 ppm pour le diol anti 53. Également, les protons en
C-2 pour le di01 80 se présentent sous forme de deux systèmes distincts (1.51
et 1.72 pprn) alors qu'ils sont regroupés en un seul multiplet (1.60 ppm) pour le
diol 53.
Schéma 33: Origine de la sélectivit4 lors de la réduction de la P-hydroxy-cetone
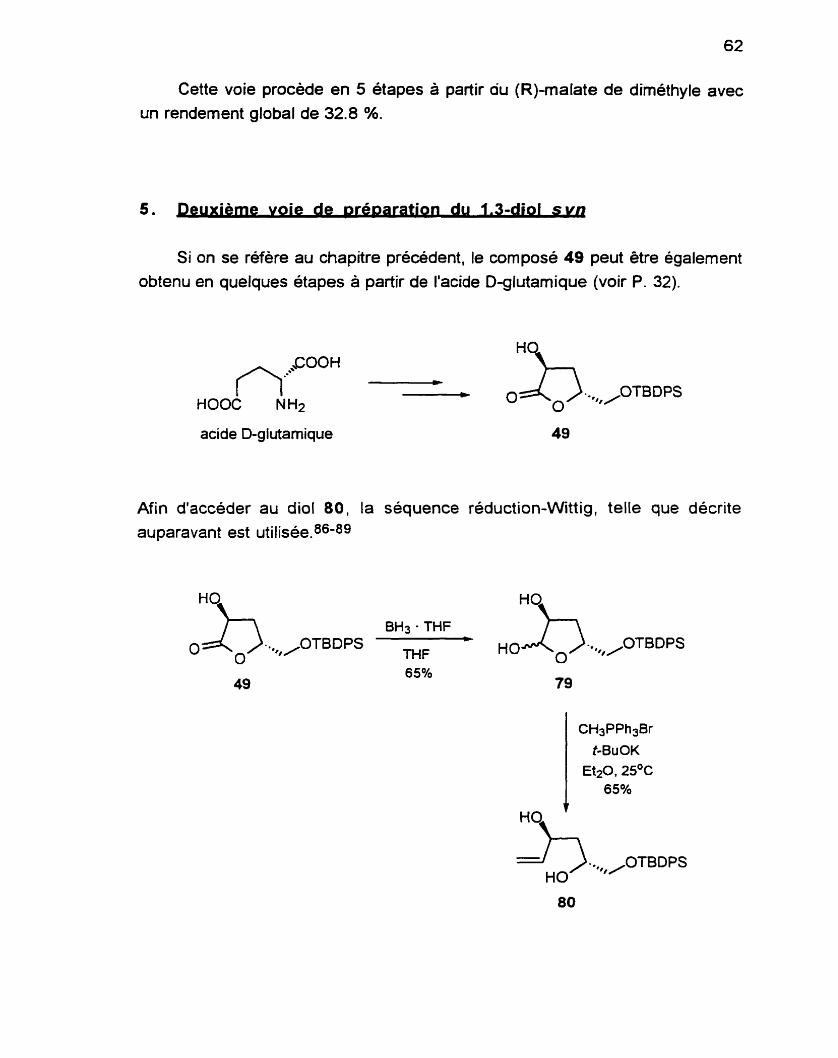
Cette voie procède en 5 étapes à partir au (R)-rnalate de diméthyle avec
un rendement global de 32.8 %.
. . 5. D e m e voie de ~ r b ~ t i o n du 1.3-diol svq
Si on se réfère au chapitre précédent, le composé 49 peut être également obtenu en quelques étapes à partir de l'acide D-glutamique (voir P. 32).
acide D-glutamique 49
Afin d'accéder au diol 80, la séquence réduction-Wittig, telle que décrite auparavant est utilisée. 86-89
EH3 - THF
' *#, 0- Ho ?-)- O ' r t / OTBDPS
.De#,/ OTBDPS

Cette séquence menant au composé 80 procède donc en 4 étapes à partir du réactif commercial 47, avec un rendement global de 26%.
6. Phase f i n u de la svnthès
Le di01 80 est mis en présence de PdC12 et de CO pour conduire au bicycle 81. dans un rendement de 90%.so9s1 Dans ce cas également, seul le bicycle de jonction cis 81 est obtenu.
PdC12, CuCI2 H
NaOAc, AcOH
%/ OTBDPS CO 0 ~ ~ 1 ~ ) 5 0 .-,,/ OTBDPS
2S°C H 90% 81
La réactivité du di01 80, par rapport à celle de son isomère 53, est la même,
soit qu'il faut compter un temps de réaction de 24 heures pour que tout le diol soit transformé.
On procède ensuite à l'élaboration de la première chaîne latérale alkyle.
/ OTBDPS -. -7
THF -78OC
,.' OTBDPS
La portion lactone est réduite par l'action du DIBAL, pour conduire au
lactol 82, avec un très bon rendement. La réaction conduit à un mélange de
diastéréoisomères et le composé peut être purifié par chromatographie sur gel de silice.
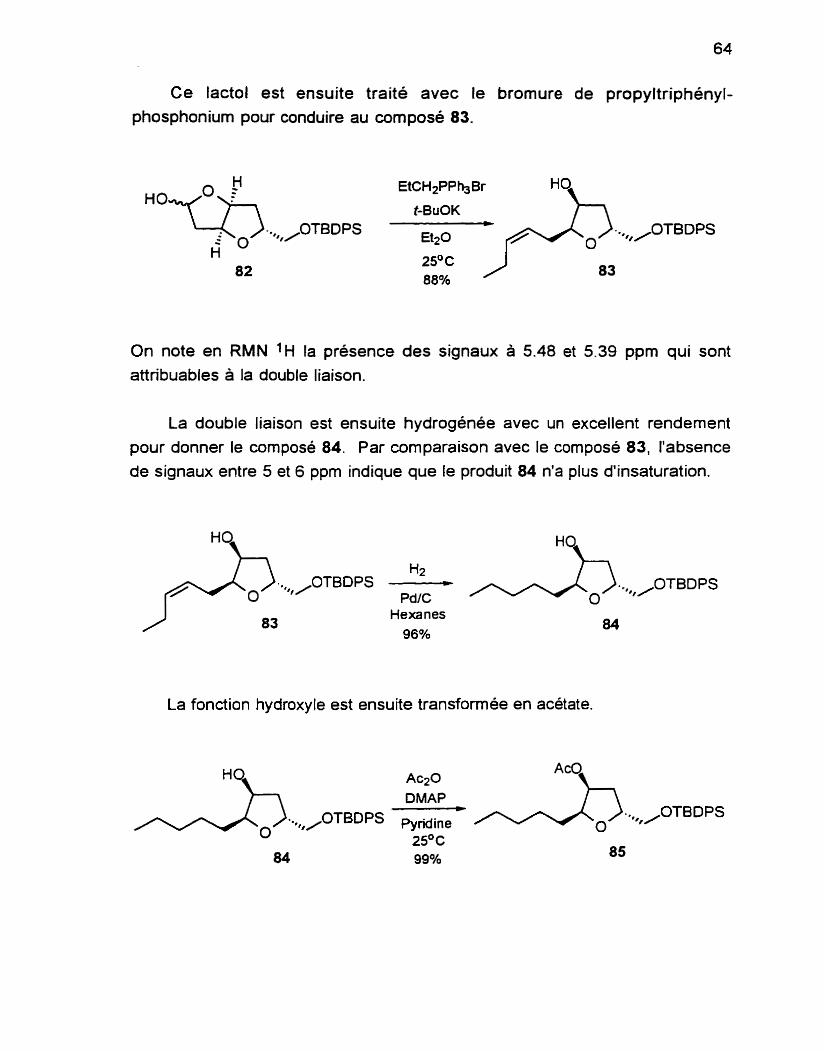
Ce lactol est ensuite traite avec le bromure de propyltriphényl- phosphonium pour conduire au composé 83.
'# t t / OTBDPS
On note en RMN r H la présence des signaux à 5.48 et 5.39 ppm qui sont attribuables à la double liaison.
La double liaison est ensuite hydrogénée avec un excellent rendement pour donner le composé 84. Par comparaison avec le composé 83, l'absence de signaux entre 5 et 6 ppm indique que le produit 84 n'a plus d'insaturation.
#*t / OTBDPS
Hexa n es 96%
OTBDPS -'#,,'
La fonction hydroxyle est ensuite transformée en acétate.
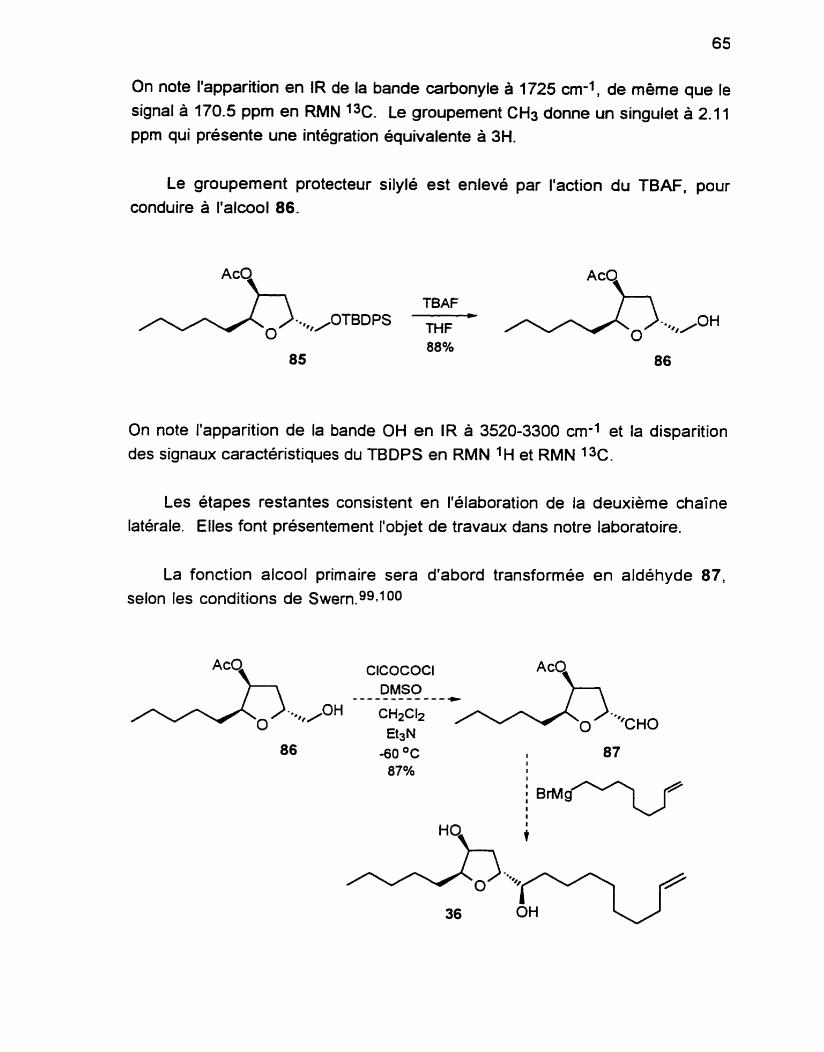
On note I'apparlion en IR de la bande carbonyle à 1725 c d , de même que le signal à 170.5 ppm en RMN 13C. Le groupement CH3 donne un singulet à 2.1 1 ppm qui présente une intégration équivalente à 3H.
Le groupement protecteur silylé est enlevé par l'action du TBAF, pour
conduire a l'alcool 86.
On note l'apparition de la bande OH en IR à 3520-3300 cm-' et la disparition des signaux caractéristiques du TBDPS en RMN l H et RMN 1%.
Les étapes restantes consistent en l'élaboration de la deuxième chaîne
latérale. Elles font présentement l'objet de travaux dans notre laboratoire.
La fonction alcool primaire sera d'abord transformée en aldéhyde 87, selon les conditions de S~ern .99~100
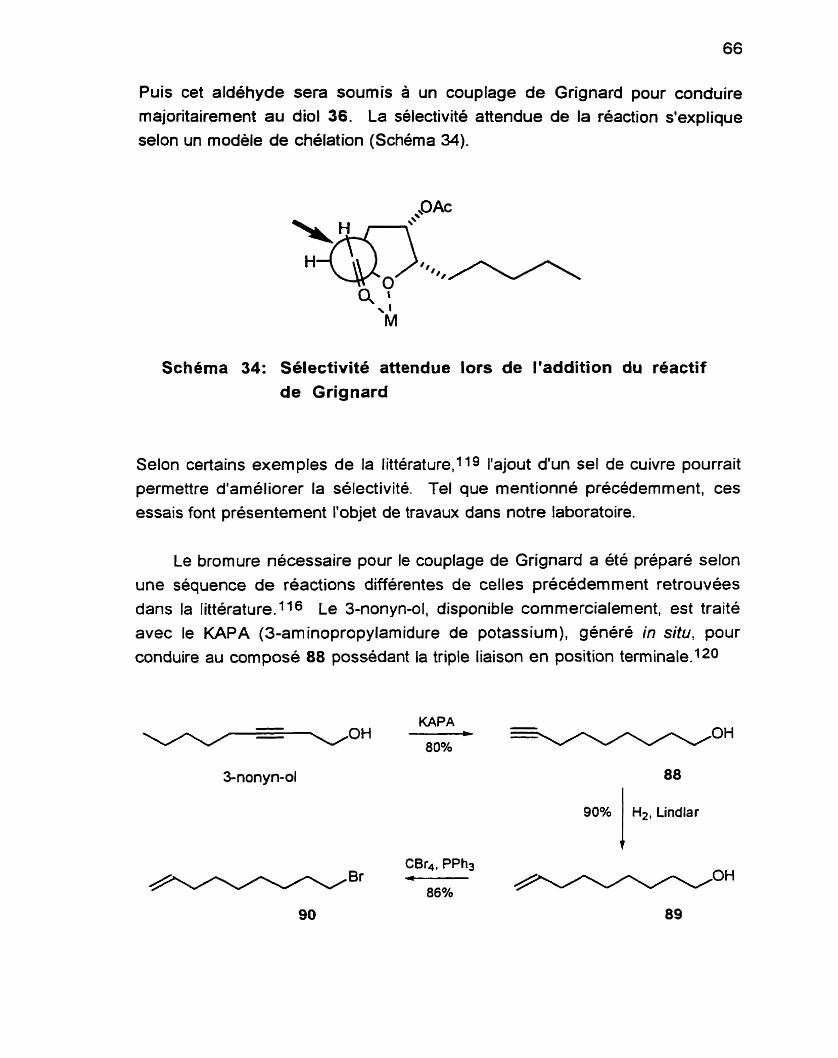
Puis cet aldéhyde sera soumis a un couplage de Grignard pour conduire majoritairement au diol 36. La sélectivité attendue de la réaction s'explique selon un modèle de chélation (Schéma 34).
Schéma 34: Sélectivité attendue lors de l'addition du réactif de Grignard
Selon certains exemples de la littérature,llg l'ajout d'un sel de cuivre pourrait permettre d'améliorer la sélectivité. Tel que mentionne précédemment, ces
essais font présentement l'objet de travaux dans notre laboratoire.
Le bromure nécessaire pour le couplage de Grignard a été préparé selon
une séquence de réactions différentes de celles précédemment retrouvées dans la littérature.116 Le 3-nonyn-01, disponible commercialement, est traité avec le KAPA (3-am inopropylamidure de
conduire au composé 88 possédant la triple
potassium), généré in situ, pour liaison en position terminale.120

Le mécanisme de cette réaction est une succession d'allènes et d'alcynes qui aboutissent à la génération d'un anion en bout de chaîne. Bien que la triple liaison interne soit favorisée thenodynamiquement, la force motrice de cette isomérisation est la formation de I'acétylure le plus stable, soit celui en bout de chaîne. L'acétylure métallique peut parfois précipiter dans le milieu réactionnel. La présence de cette triple liaison est confirmée par le signal à 1.91 ppm en RMN 'H et par l'absence en IR de bandes vers 2100 cm-1 qui sont attribuables à une triple liaison interne. Les données spectroscopiques pour ce composé sont en accord avec celles retrouvées dans la littérature.121 La triple liaison est ensuite réduite sélectivement en double liaison à l'aide du catalyseur de Lindlar (Pd 1 C ~ C O ~ ) . ~ Z En RMN ' H, la disparition du signal à
1.91 ppm et la présence de signaux à 5.79 et 4.95 ppm (CHz=CH-) confirme l'identité du produit obtenu, soit l'alcène 89.123 La fonction hydroxyle est finalement transformée en bromure 90.124 Les données spectroscopiques
pour ce composé sont également en accord avec celles retrouvées dans la
littérature.
Cette séquence vers le di01 36 devrait donc procèder en 12 étapes a partir
du réactif commercial 47.
Cette stratégie de synthèse devrait aussi permettre l'accès aux autres membres de cette famille de tétrahydrofuranes (Schéma 35).
%, / OTBDPS ------' '"8, / OTBDPS
Schéma 35: Accès aux autres tétrahydrofuranes

En effet, on pourra accéder aux composes 71 -78 par manipulation judicieuse
des groupements protecteurs des fondions hydroxyles.

CHAPITRE 3
Synthese totale du plakortone D
Les éponges marines (Phylum Porifera) sont considérées comme des organismes multicellulaires primitifs, possédant une organisation interne relativement simple. Ces éponges, particulièrement celles qui n'ont pas de
spicules, produisent de grandes quantités de métabolites secondaires qui ont notamment pour fonction de repousser les prédateurs potentiels et d'inhiber la
croissance d'organismes parasitaires.
Ces espèces ont reçu beaucoup d'attention de la part des chercheurs,
chimistes et biochimistes, qui les ont étudiées principalement pour les mécanismes de défense chimique que ces organismes, peu mobiles, fragiles et physiquement sans protection, déploient.
Les espèces du genre Plakortis se sont particulièrement distinguées comme une source de composés ayant des propriétés biologiques intéressantes. Ces éponges contiennent entre autres des peroxydes cycliques
(par exemple, les composés 91126 et 92127) et d'autres composés apparentés qui ont démontré des propriétés anti-cancer et anti-fongiques.
Mais ce qui se revele l'activité la plus intéressante d'un nombre significatif de ces composés est sans nul doute leur potentiel d'activation des ATPases pompant les ions Ca2+ dans le réticulum sarcoplasrnique des membranes. Le
retrait rapide des ions Ca2+ du cytosol, indispensable à la relaxation des cellules du muscle cardiaque, est effectuée principalement par ces ATPases. Une rétention altérée de ces ions Ca2+ par le réticulum sarcoplasmique cardiaque peut contribuer a la relaxation anormale observée dans certains cas
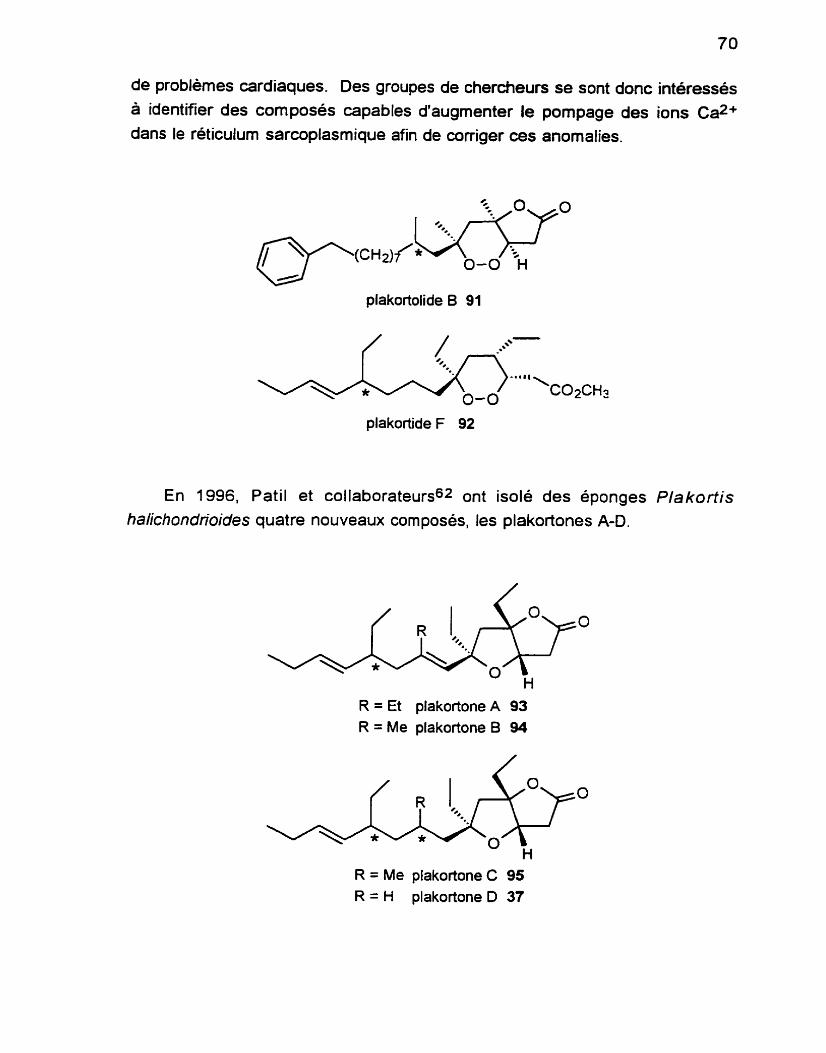
de problèmes cardiaques. Des groupes de chercheurs se sont donc intéressés à identifier des composés capables d'augmenter le pompage des ions Ca2+ dans le réticulum sarcoplasmique afin de corriger ces anomalies.
plakortolide B 91
plakortide F 92
En 1996, Patil et collaborateurs62 ont isolé des éponges Plakorfis halichonddoides quatre nouveaux composés, les plakortones A-D.
'8
O H
R = Et plakortone A 93 R =Me plakortone B 94
R = Me plakortone C 95 R = H plakortone D 37
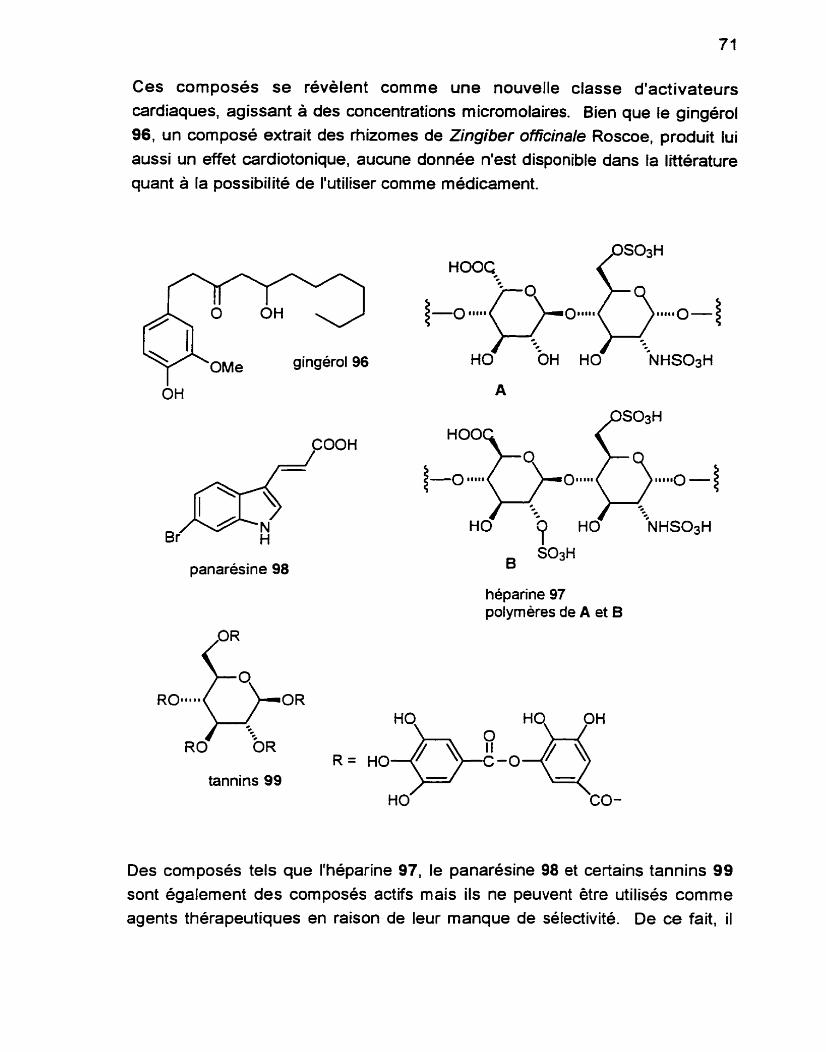
Ces composés se révèlent comme une nouvelle classe d'activateurs
cardiaques. agissant à des concentrations micromolaires. Bien que le gingérol 96, un composé extrait des rhizomes de Zingiber officinale Roscoe, produit lui
aussi un effet cardiotonique, aucune donnée n'est disponible dans la littérature quant à la possibilité de l'utiliser comme médicament.
panarésine 98
tannins 99
héparine 97 polymères de A et B
Des composés tels que l'héparine 97, le panarésine 98 et certains tannins 99
sont également des composés actifs mais ils ne peuvent être utilisés comme agents thérapeutiques en raison de leur manque de sélectivité. De ce fait, il

n'existe a ce jour aucun composé satisfaisant pour activer les ATPases pompant les ions Ca2+ dans le muscle ~ardiaque-~z Les plakortones A-D sont donc une piste fort prometteuse vers la découverte d'un médicament adéquat
pour le traitement de certaines maladies cardiaques.
Les plakortones se distinguent par une structure bicyclique de type furane-
furanone sur laquelle est greffée une chaîne latérale aliphatique ramifiée. La détermination de ces structures a été faite sur la base de données spectrales. La stéréochimie relative de l'unité bicyclique a été résolue à l'aide d'expériences NOE. Cependant, la stéréochimie sur les chaînes latérales n'a pu être déterminée ce qui implique que la stéréochimie absolue de ces composés n'est pas connue. La synthèse totale de ceux-ci permettrait ainsi de résoudre ce problème.
À ce jour, aucune synthèse de ces composés n'a encore été rapportée
dans la littérature. Le choix d'une cible s'est porté sur le plakortone D car dans ce cas, il n'y a qu'un seul centre stéréogénique à contrôler sur la chaîne latérale. De plus, le plakortone D s'avère le plus actif de tous les membres de cette famille.
L'observation de la structure du composé-cible nous révèle deux principales sous-unités, soient la chaîne latérale et le bicycle (Schéma 36). Nous avons envisagé de préparer ces deux composantes et de les réunir par un couplage de type Wittig. Puisque la stéréochimie de la chaîne latérale n'est pas connue, on devra réaliser la synthese des deux antipodes de cette dernière. Le bicycle proviendra d'une réaction d'alkoxycarbonylation- lacton isation catalysée par le Pd( l l),soe51 telle que réalisée dans les chapitres précédents. II est à noter que dans ce cas, le 1,3-diol précurseur à cette transformation devra posséder deux alcools tertiaires optiquement actifs. Les sections qui suivent présentent les voies de synthese utilisées pour la préparation et le couplage de ces synthons.
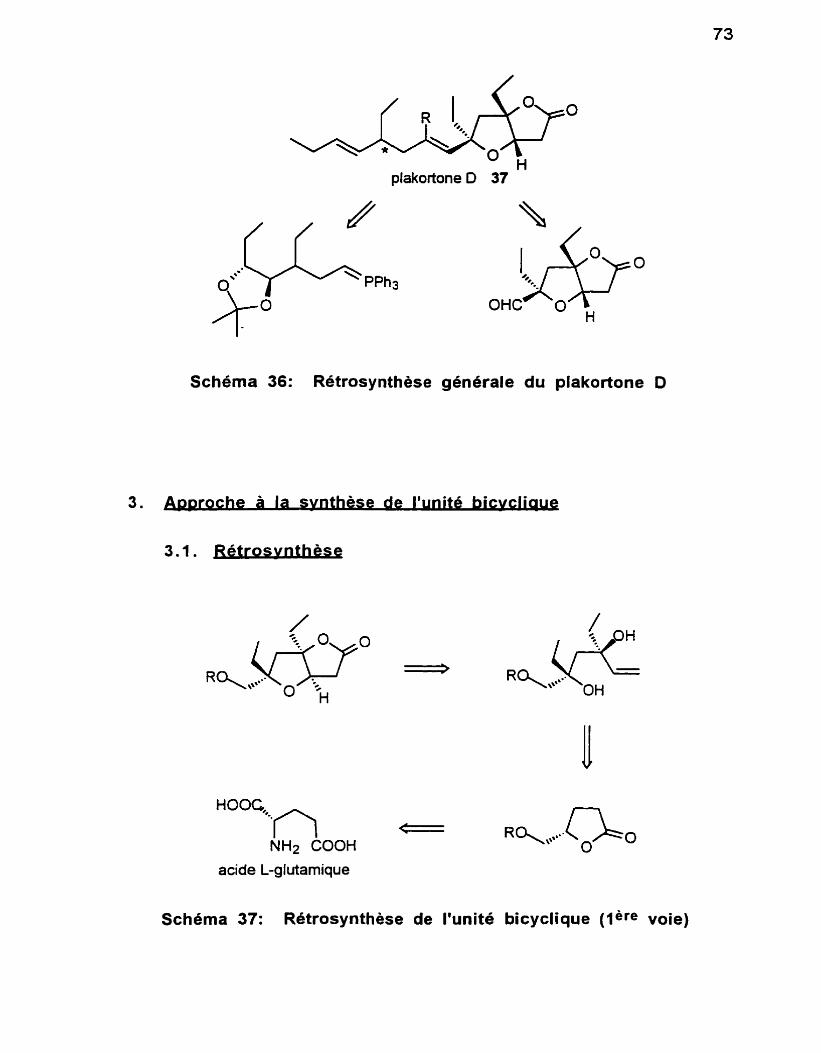
- .
plakortone D 37
OHC H
Schéma 36: Rétrosynthèse gén6rale du plakortone D
acide L-glutamique
Schéma 37: Rétrosynthèse de l'unité bicyclique ( le '= voie)
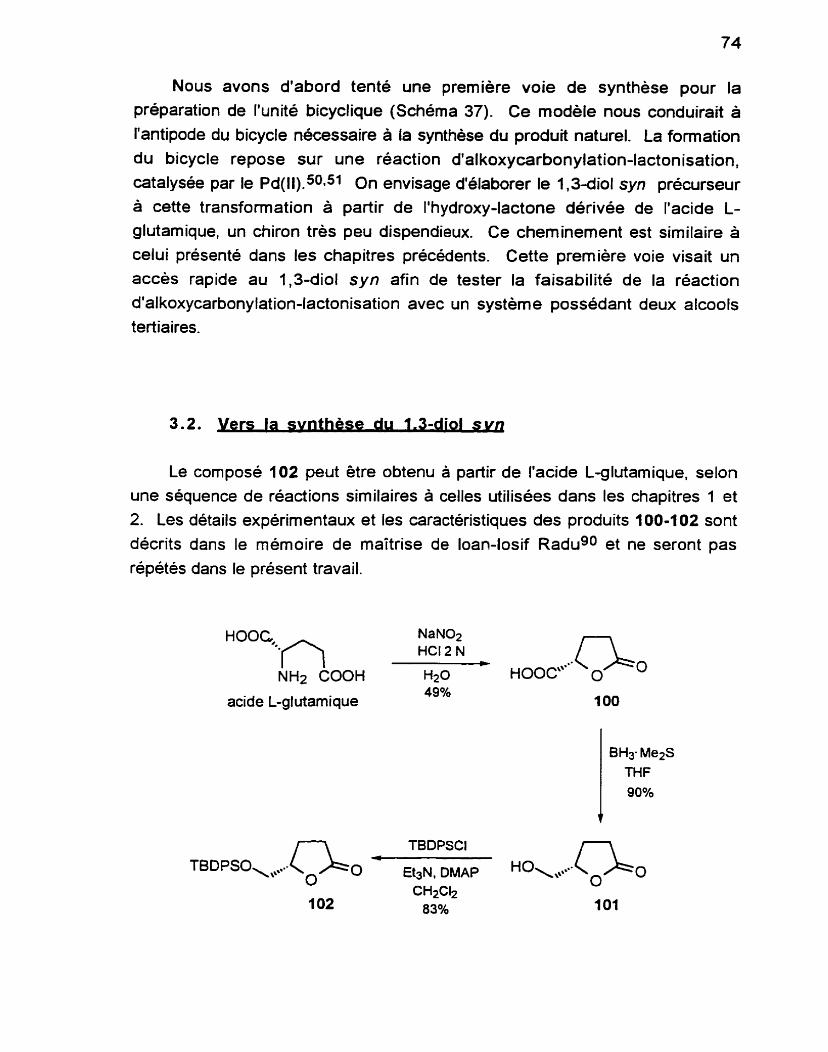
Nous avons d'abord tenté une première voie de synthèse pour la
préparation de l'unité bicyclique (Schéma 37). Ce modèle nous conduirait a
l'antipode du bicycle nécessaire à la synthèse du produit naturel. La formation du bicycle repose sur une réaction d'alkoxycarbonylation-lactonisation, catalysée par le ~d(11).50~51 On envisage d'élaborer le l,3-diol syn précurseur à cette transformation à partir de I'hydroxy-lactone dérivée de l'acide L- glutamique, un chiron très peu dispendieux. Ce cheminement est similaire à celui présenté dans les chapitres précédents. Cette première voie visait un accès rapide au 1.3-diol syn afin de tester la faisabilité de la réaction d'alkoxycarbonylation-lactonisation avec un système possédant deux alcools tertiaires.
3.2. Vers la svnthèse du 1 -3-diol s v ~
Le composé 102 peut être obtenu a partir de l'acide L-glutamique, selon une séquence de réactions similaires à celles utilisées dans les chapitres 1 et 2. Les détails expérimentaux et les caractéristiques des produits 100-1 02 sont
décrits dans le mémoire de maitrise de loan-losif Radugo et ne seront pas répétés dans le présent travail.
HooCn NaNO2 HCI 2 N
* NHz COOH H20
49% acide L-gl utamique
TBDPSCI
TBDPSO,,,,... 0 0 n 'i Et& DMAP
BH3- Me2S THF 90%
1
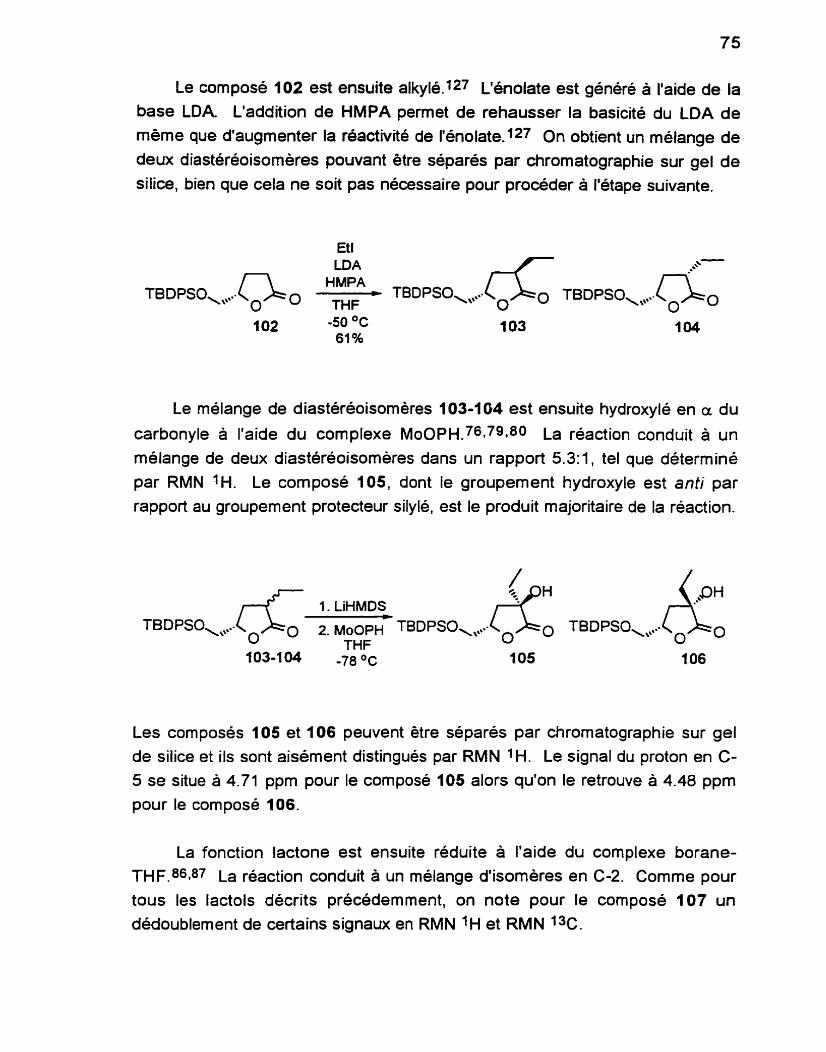
Le composé 102 est ensuite alkylé.127 L'enolate est généré à I'aide de la base LDA L'addition de HMPA permet de rehausser la basicité du LDA de même que d'augmenter la réactivité de l'énolate.127 On obtient un mélange de deux diastéréoisomères pouvant être séparés par chromatographie sur gel de
silice, bien que cela ne soit pas nécessaire pour procéder à l'étape suivante.
Etl LDA \-
HMPA TB DPSO,,,... a, 7 TBDPSO,,,..-
O O TBDPSO,,,...
O
Le mélange de diastéréoisomères 103-104 est ensuite hydroxylé en a du
carbonyle à I'aide du complexe MoOPH.76.79.80 La réaction conduit à un mélange de deux diastéréoisomères dans un rapport 5.3:1, tel que déterminé
par RMN 1H. Le composé 105, dont le groupement hydroxyle est anfi par rapport au groupement protecteur silylé, est le produit majoritaire de la réaction.
V - u - THF - u -
Les composés 105 et 106 peuvent être séparés par chromatographie sur gel de silice et ils sont aisément distingués par RMN 1H. Le signal du proton en C- 5 se situe a 4.71 ppm pour le composé 105 alors qu'on le retrouve à 4.48 ppm pour le composé 106.
La fonction lactone est ensuite réduite à I'aide du complexe borane- THF.86.87 La réaction conduit à un mélange d'isomères en C-2. Comme pour tous les lactols décrits précédemment, on note pour le composé 107 un
dédoublement de certains signaux en RMN 1H et RMN 136.
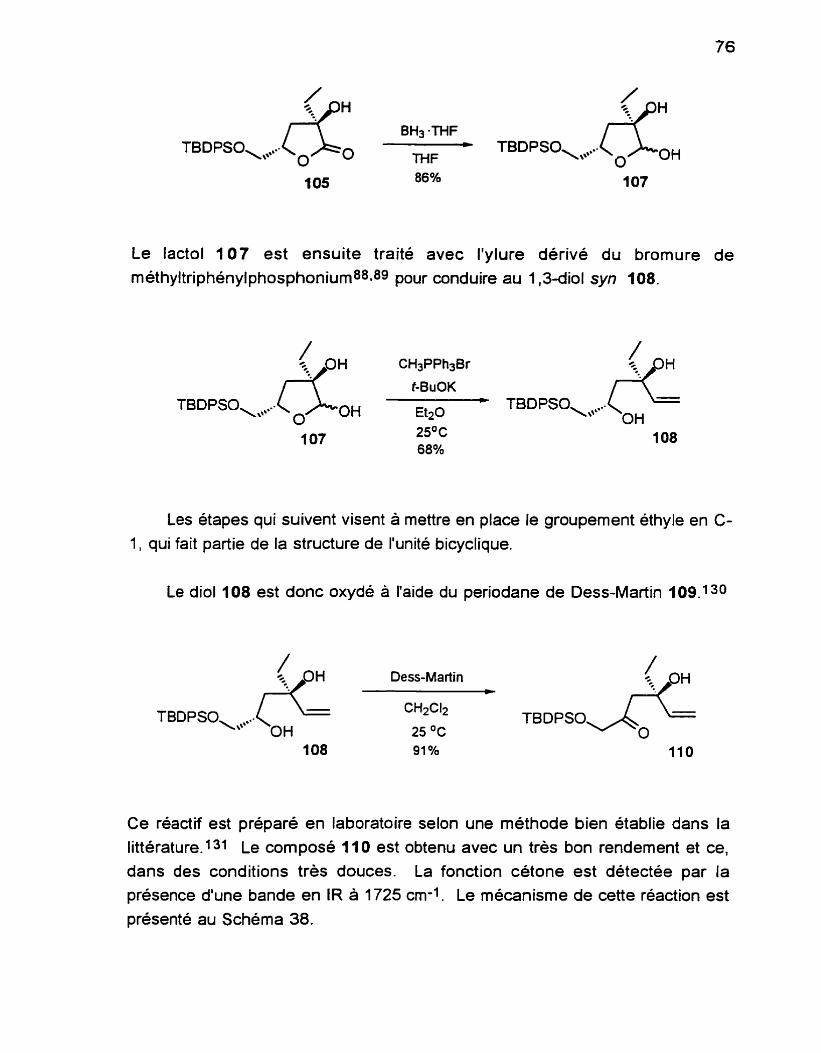
Le lactol 107 est ensuite traité avec I'ylure dérivé du bromure de
méthyltriphénylpho~phonium8*~89 pour conduire au 1,3-di01 syn 108.
Les étapes qui suivent visent à mettre en place le groupement éthyle en C- l, qui fait partie de la structure de l'unité bicyclique.
Le di01 108 est donc oxydé à l'aide du periodane de Dess-Martin 109.130
/ Dess-Martin /
& - CH2CI2
TBDPSO,,,,. .- TBDPSO OH 25 OC
1 08 91 % d 11 0
Ce réactif est préparé en laboratoire selon une méthode bien établie dans la littérature.131 Le composé 110 est obtenu avec un très bon rendement et ce, dans des conditions très douces. La fonction cétone est détectée par la présence d'une bande en IR à 1725 cm-'. Le mécanisme de cette réaction est
présenté au Schéma 38.
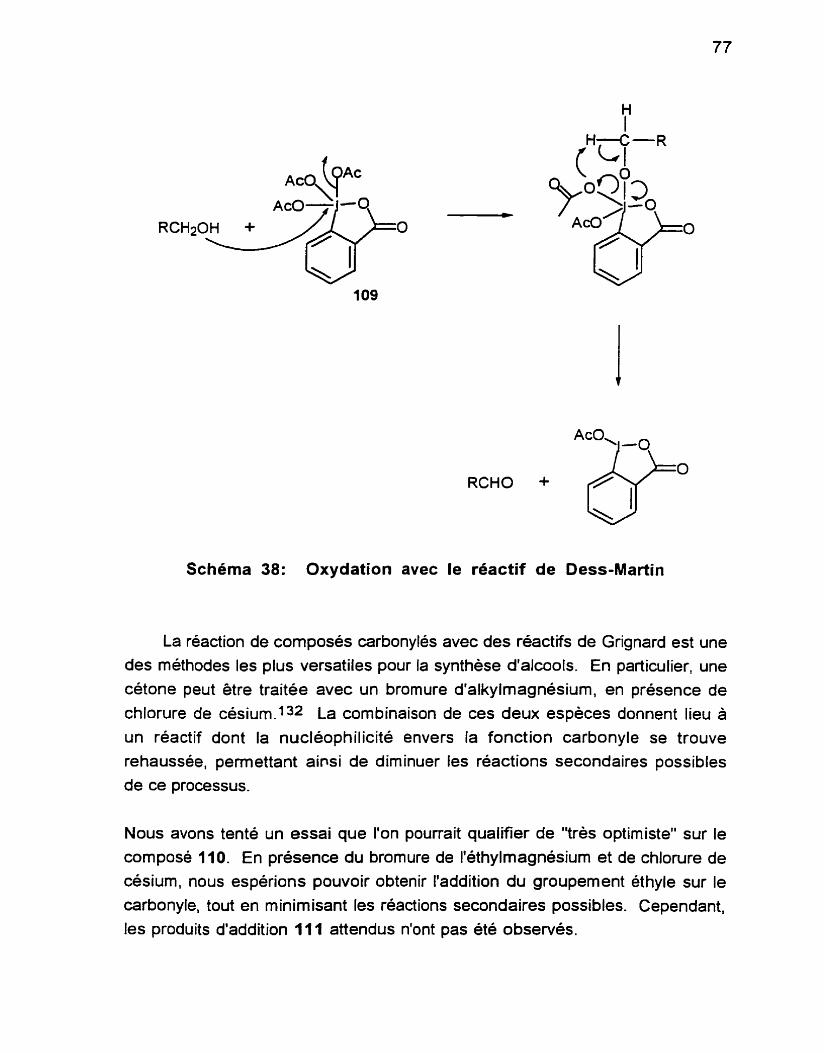
AcO, 1-4
Schéma 38: Oxydation avec le réactif de Dess-Martin
La réaction de composés carbonylés avec des réactifs de Grignard est une
des méthodes les plus versatiles pour la synthèse d'alcools. En particulier, une
cétone peut être traitée avec un bromure d'alkylmagnésium, en présence de
chlorure de césium.132 La combinaison de ces deux espèces donnent lieu a
un réactif dont la nucléophilicité envers la fonction carbonyle se trouve rehaussée, permettant ainsi de diminuer les réactions secondaires possibles de ce processus.
Nous avons tenté un essai que l'on pourrait qualifier de ''très optimiste" sur le
composé 11 0. En présence du bromure de I'ethylmagnésium et de chlorure de césium, nous espérions pouvoir obtenir l'addition du groupement éthyle sur le carbonyle. tout en minimisant les réactions secondaires possibles. Cependant,
les produits d'addition 11 1 attendus n'ont pas été observés.
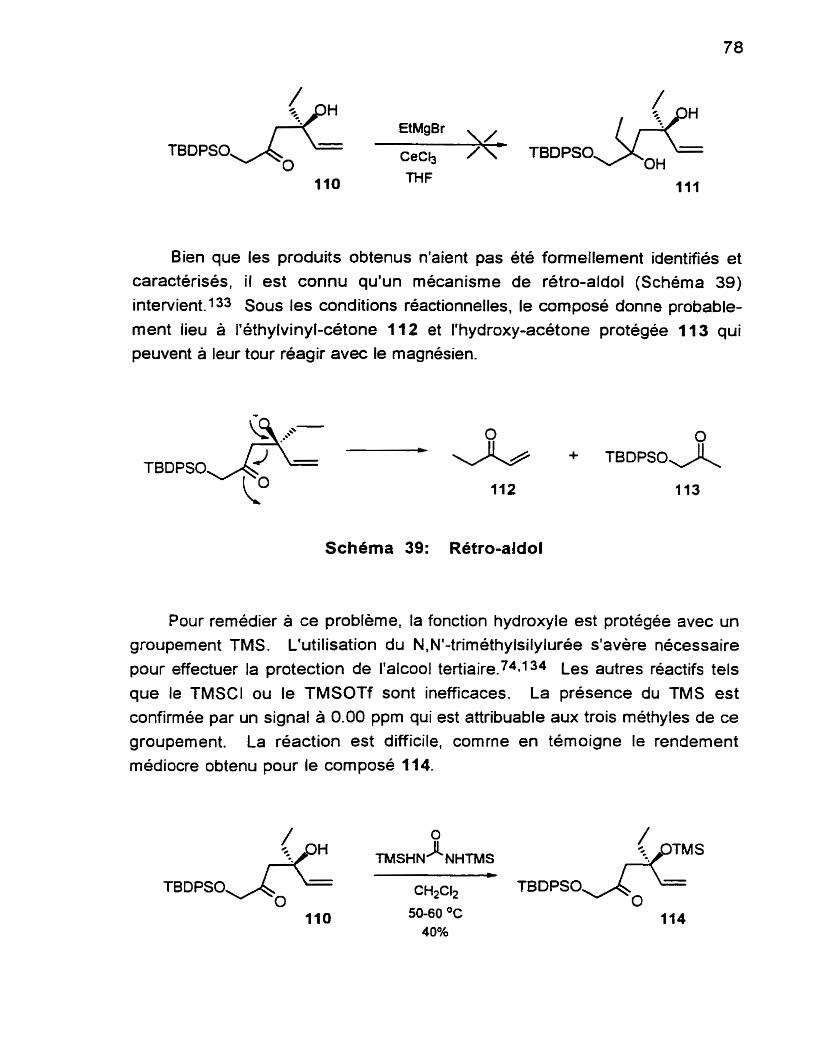
THF
Bien que les produits obtenus n'aient pas été formellement identifiés et caractérisés, il est connu qu'un mécanisme de rétro-aldol (Schéma 39)
intervient.133 Sous les conditions réactionnelles, le composé donne probable-
ment lieu à I'éthylvinyl-cétone 1 12 et Ithydroxy-acétone protégée 1 13 qui
peuvent à leur tour réagir avec le magnésien.
Schéma 39: Rétro-aldol
Pour remédier a ce problème, la fonction hydroxyle est protégée avec un groupement TMS. L'utilisation du N, Nt-triméthylsilylurée s'avère nécessaire pour effectuer la protection de l'alcool te^-tiaire.74J34 Les autres réactifs tels que le TMSCl ou le TMSOTf sont inefficaces. La présence du TMS est confirmée par un signal à 0.00 ppm qui est attribuable aux trois méthyles de ce
groupement. La réaction est difficile, cornrne en témoigne le rendement
médiocre obtenu pour le composé 114.
K TMSHN NHTMS

Malheureusement, la réaction du composé avec le bromure d'éthylmagnésium
en présence de chlorure de césium ne permet pas d'obtenir le produit 11 5 attendu.
THF
Face à ces insuccès à obtenir le 1,3-diol tertiaire précurseur, nous avons entrepris l'étude d'une nouvelle voie pour la préparation du bicycle. Elle sera présentée dans la prochaine section.
II est cependant important de noter qu'il existe dans la littérature une autre
alternative à l'utilisation d'un Grignard pour l'addition d'un groupement alkyle ou aryle sur un composé de type p-hydroxycétone (Schéma 40).
Schéma 40: Addition de l'espèce RLi-CeC13
La combinaison d'un organolithien avec le chlorure de césium donne lieu
également à une espèce qui permet le transfert d'un groupement vers le carbonyle pour conduire au 1,3-di01 avec un excellent rendernent.133 La réaction fonctionne bien avec une variété de groupements et les produits obtenus sont des 1.3-diols bis-tertiaires, sans que des produits de réactions
secondaires ne soient détectés. Les auteurs travaillent présentement à élargir
cette méthode à la préparation de composés énantiornériquernent purs.

Nous n'avons pas entrepris cette ultime tentative puisque la voie de synthèse depuis l'acide L-glutam ique a été jugée trop la borieuse.
Mais auparavant, une dernière expérience concernant cette voie a été réalisée. Le diol 108 a été soumis aux conditions d'alkoxycarbonylation- lactonisation.50.51 II s'agissait de vérifier si le groupement éthyle en C-3 pouvait nuire à la réaction, et ce, même si des exemples de la littérature démontrent que la réaction procède sans difficultés.so~51
Le bicycle 116 est obtenu comme seul et unique produit, avec un rendement non optimisé de 61%. Ce résultat était fort encourageant et il était ainsi
possible de croire que le deuxième centre quaternaire n'affecterait pas le
déroulement de la cyclisation.
4. Svnthè I ' se de 1 unité b
Tel que précédemment, le bicycle provient d'une réaction d'alkoxy-
carbonylation-lactonisation5O151 catalysée par le Pd(ll) (Schéma 41). Dans ce cas, le 1,3-diol syn précurseur à cette transformation provient d'une méthylénation sur le lactol dérivé d'une butyrolactone substituée. Cette
dernière est obtenue par ouverture puis lactonisation subséquente d'un époxyde connu. II est important de mentionner à nouveau que le 1,3-diol syn possède deux alcools tertiaires et qu'il existe très peu de méthodologies pour préparer un tel synthon.
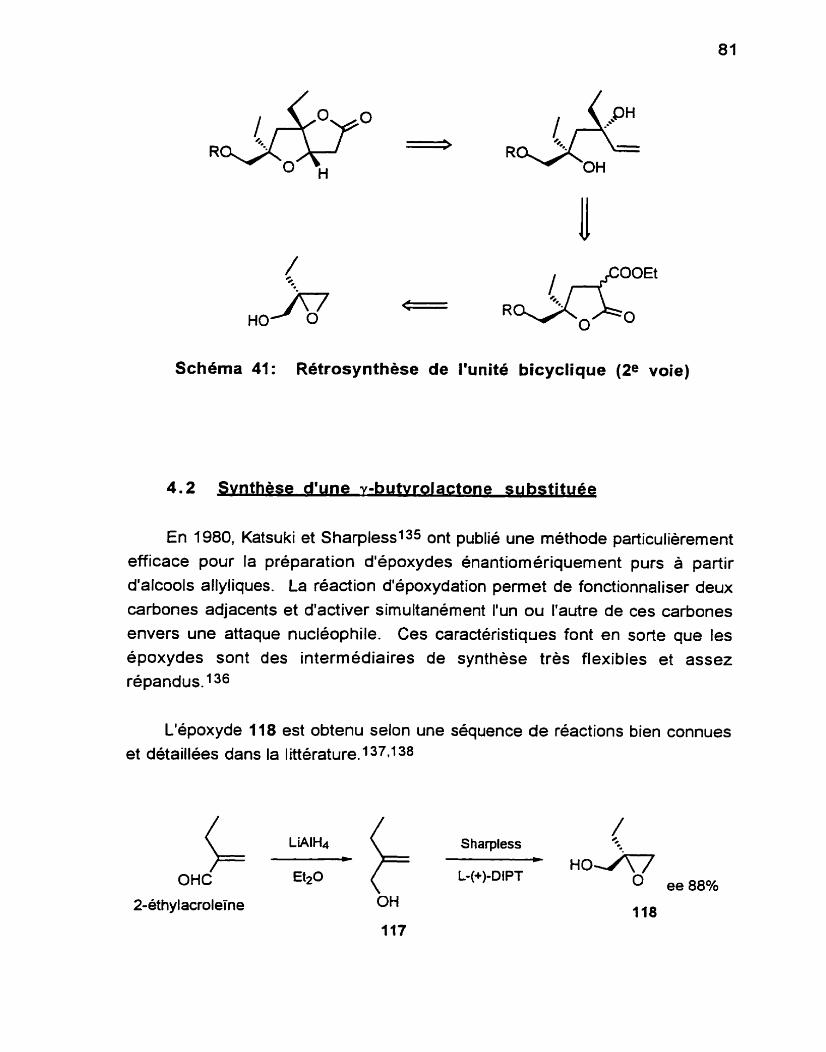
Schéma 41 : Rétrosynthèse de l'unité bicyclique (2e voie)
4.2 Svnthèse d'me Y-butvrolactone substituée
En 1980, Katsuki et Sharplessl3s ont publié une méthode particulièrement efficace pour la préparation d'époxydes énantiomériquement purs à partir
d'alcools allyliques. La réaction d'époxydation permet de fonctionnaliser deux
carbones adjacents et d'activer simultanément l'un ou l'autre de ces carbones
envers une attaque nucléophile. Ces caractéristiques font en sorte que les époxydes sont des intermédiaires de synthèse très flexibles et assez
répandus. 36
L'époxyde 118 est obtenu selon une séquence de réactions bien connues
et détaillées dans la littérature? 37,138
2-éthylacro leïne OH 118

L'excès énantiomère a été déterminé par RMN rH sur le dérivé de Mosher.139 On note en RMN I H les doublets a 2.86 et 2.66 ppm qui sont caractéristiques des protons de I'oxirane.
II est important de noter que la réaction de Sharpless doit être réalisée rigoureusement selon les directives données. Le facteur le plus crucial de cette réaction semble être la solution de TBHP.136a Une solution 5-6M dans du 2,2,4-triméthylpentane était autrefois disponible commercialement et permettait l'obtention de très bons résultats pour I'époxydation du composé 117. Des essais avec une solution commerciale dans du décane se sont avérés désastreux. La réaction ne fonctionnait tout simplement pas. Sharpless et ~ohnson136a recommandent l'utilisation d'une solution commerciale dans de I'isooctane mais banissent toute autre solution disponible commercialement. II
est également possible de préparer la solution de TBHP dans du toluène ou du CH2CI2.I40 II faut alors veiller à ce que la concentration de la solution se situe entre 4 et 6 M pour de meilleurs résultats. Une solution de 3M par exemple s'avère trop diluée et le solvant pourrait alors avoir un effet inhibiteur sur le
déroulement de la réaction.
L'époxy-alcool 118 est protégé à l'aide du groupement TBDPS, avec un bon rendement.
Et3N, DMAP - _. TBDPSU-/'
O CH2C12 O
L'époxyde 11 9 est ensuite ouvert avec l'anion du malonate de diéthyle.141 Durant le traitement acide du milieu réactionnel, il y a lactonisation pour conduire au composé 120, avec un excellent rendement, sous forme d'un mélange d'isomères. La réaction nécessite des conditions drastiques pour parvenir à ouvrir cet époxyde qui n'est pas activé. Le produit obtenu est un mélange qu'il n'est pas nécessaire de séparer pour l'étape suivante.
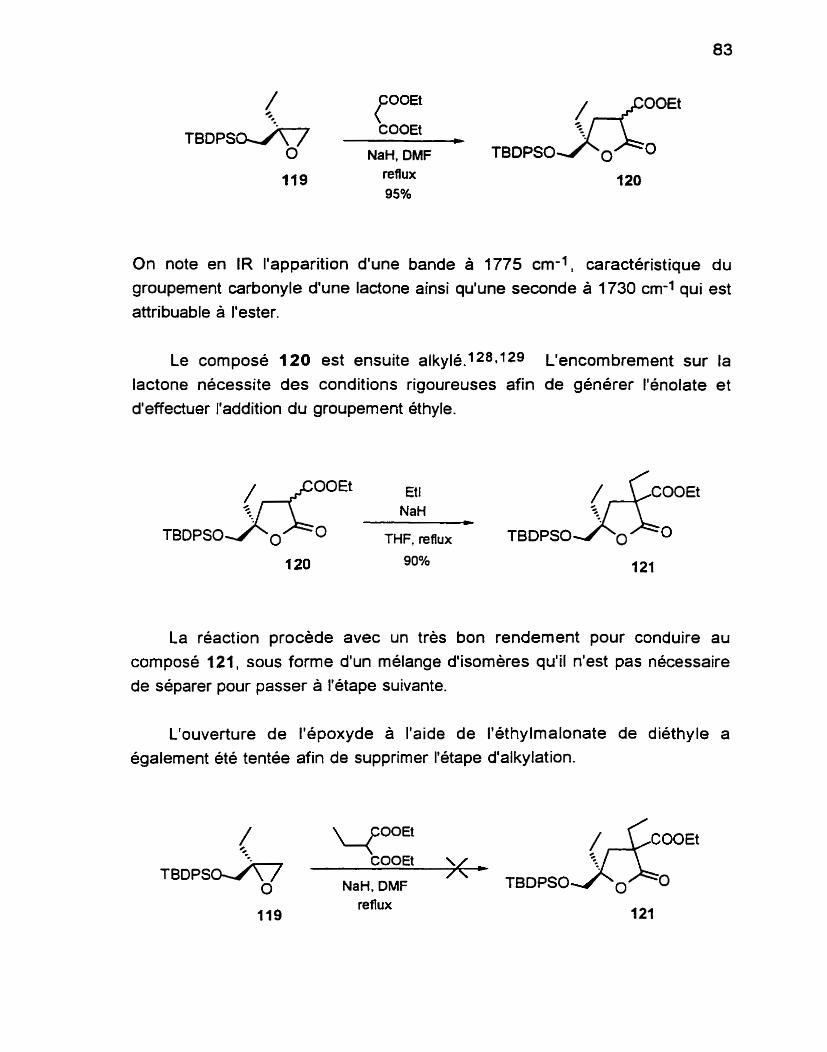
/ #
5
T B D P S ~ ~ [*OEt COOEt
;c
0 NaH, DMF TBDPSO
11 9 reflux 95%
On note en IR l'apparition d'une bande à 1775 cm-', caractéristique du groupement carbonyle d'une lactone ainsi qu'une seconde à 1730 cm-' qui est
attribuable à l'ester.
Le composé 1 20 est ensuite alky1é.12~912~ L'encombrement sur la lactone nécessite des conditions rigoureuses afin de générer I'énolate et
d'effectuer l'addition du groupement éthyle.
TBDPSO
Etl NaH
THF, reflux TBDPSO O
La réaction procède avec un très bon rendement pour conduire au composé 121, sous forme d'un mélange d'isomères qu'il n'est pas nécessaire
de séparer pour passer à l'étape suivante.
L'ouverture de l'époxyde à l'aide de I'éthylmalonate de diéthyle a
également été tentée afin de supprimer l'étape d'alkylation.
reflux

Cependant, le composé 121 n'est pas obtenu dans ces conditions. La génération de I'énolate est visible (la suspension de NaH devient limpide lorsque ce dernier est entièrement consommé) mais le réactif doit être trop volumineux pour réagir avec l'époxyde, qui lui aussi est encombre.
Le composé 121 est ensuite décarboxylé en présence de Li&, à reflux dans le DMF.141
LiBr -
TBDPSO +
DMF, reflux TBDPSO
Le composé 122 est obtenu avec un excellent rendement, sous forme d'un
mélange d'isomères. Dans ce cas également, il n'est pas nécessaire de
séparer les composés pour passer à l'étape suivante. En IR, on note la disparition de la bande à 1725 cm-1 qui était caractéristique du groupement
carbonyle de l'ester.
L'ouverture de I'époxyde a également été tentée à l'aide de l'acide butyrique afin d'obtenir directement le composé 1 22. L'étape de
décarboxylation aurait ainsi été éliminée.
l #. CH3CH2CH2COOH
T B D P Ç ~ ~ LDA 4
0 THF:HMPA (6:l) TBDPSO
reflux '1 22
Cependant, le composé 122 est obtenu avec un médiocre rendement dans ces conditions. II s'avère donc plus efficace d'utiliser la séquence de réactions

(ouverture de l'époxyde, alkylation et décarboxylation) telles que décrites précédemment.
Le composé 122 est ensuite hydroxylé. Les essais avec le
M O O P H ~ ~ ~ ~ ~ ~ * ~ ont conduit à une médiocre sélectivité (Voir Tableau 1). II s'avérait alors nécessaire de trouver un autre réactif qui permettrait d'obtenir une meilleure sélectivité pour le composé 123.
TBDPSO
1. Base
2. "Agent hydroxylant" TBDPSO
THF
f BDPSO
Agents Base 1 2 3 1 2 4 hydroxylants
125 NaHMDS 1.2 1
126 NaHMDS 6.0 1
Tableau 1: Sélectivité des essais d'hydroxylation
En 1986, Davis et collaborateurs14* ont introduit une nouvelle classe d'oxaziridines énantiomériquement pures permettant I'hydroxylation

asymétrique d'énolates avec de très bonnes s6lectivlés. Les réactifs 125 et 126 sont disponibles commercialement ainsi que leurs énantiomères respectifs.
Le composé 127 de même que son énantiomère peuvent être préparés selon une méthode bien detaillée.143 C'est la configuration de I'oxaziridine qui détermine la stéréochimie du produit final. Le choix de l'énantiomère pour la transformation du composé 123 a été fait sur la base du modèle de la 2-méthyl- y-butyrolactone étudiée par Davis (Schéma 42).144 Le réactif 126 permet
d'obtenir une meilleure sélectivité en faveur du composé 123, avec un rendement de 65%. Toutes les sélectivités des hydroxylations ont été mesurées après chromatographie (masse des composés recueillis). Cependant, la sélectivité de la réaction avec le réactif 126 a également été déterminée par H P L C ' ~ ~ et se situe à plus de 10:l. Nous avons tenté de prouver la stéréochimie relative des composés obtenus par des experiences NOE mais il s'avère impossible à ce stadeci de différencier sans ambiguité les deux groupements éthyles de ces molécules. La preuve de stéréochimie sera donnée a une étape subséquente.
3) R-CI
Schéma 42: Hydroxylation de la 2-m6thylr-butyrolactone

Plusieurs exemples sont connus dans la littérature qui décrivent l'enrichissement énantiomérique de composés non-racémiques et énantio- meriquement irn purs. 46 Ces composés peuvent être enrichis par recristal- lisation, sublimation ou encore chromatographie.
Dans certains cas, une transformation chimique spécifique peut conduire à un tel résultat. La réaction d'a-hydroxylation décrite précédemment permet
de rehausser I'énantiosélectivité initiale obtenue lors de I'époxydation. On suppose que le centre stéréogénique en C-5 n'a aucun effet sur le déroulement de la réaction. Le raisonnement de ce processus se décrit comme suit:
FI- ee = 86*h, soit 9317
TBDPS O
TBDPS ONa
/
TBDPS
Schéma 43: Amplification de I'énantios6lectivit6 initiale
Afin de faciliter les calculs, nous allons supposer que l'excès énantiomère du compose 119 est de 86%, soit un rapport de 937 (Schéma 45). La réaction
d'hydroxylation sur le composé 122 conduit à une sélectivité d'au moins 10:l

en faveur de l'alcool de configuration S. En tenant compte de l'excès énantiomère du composé de départ, ceci donne lieu aux proportions indiquées pour les composés 123, 124, ent-123 et ent-124. Les composés 124 et ent-124 sont des énantioméres, ce qui implique qu'ils se retrouvent ensembles lors de la séparation sur gel de silice. Les composés 123 et ent- 123 sont forcément eux aussi des énantiomères. Si on résout l'équation suivante:
On trouve alors que x = 0.975 ce qui correspond à un excès énantiomère de 97.5%.
4.3 Svnthèse du 1.3-di01 svq
Le composé 123 est ensuite réduit en présence de DIBAL. Contrairement aux exemples présentés dans les chapitres précédents, la réaction fonctionne avec ce réactif, même si la fonction hydroxyle est libre.*' La présence du
groupement éthyle sur le C-3 doit avoir pour effet d'empêcher la complexation
du réactif avec le OH libre.
DlBAL t
TBDPSO O TBDP SO OH THF -78OC
Le lactol 128 est obtenu quantitativement sous forme d'un mélange d'isomères et il peut être purifié aisément par chromatographie sur gel de silice.
Le lactol 128 est ensuite traité avec I'ylure dérivé du bromure de rnéthyltriphenylpho~phonium*8~89 pour conduire au 1'3-diol syn 129.

Contrairement aux exemples précédents, l'encombrement sur la molécule impose le reflux dans l'éther pour permettre l'obtention du composé.
reflux
En RMN 1H, les signaux à 5.68, 5.22 et 4.89 ppm sont caractéristiques de la double liaison terminale.
Le synthon 129 possède des particularités structurales qu'il s'avère important de souligner. Ce composé est un 1,3-di01 syn optiquement actif et de plus, les deux centres stéréogéniques sont quaternaires.
Les alcools tertiaires et leurs dérivés sont des synthons très utiles pour la
préparation de nombreux médicaments et produits naturels tels que l'analogue de la prostaglandine 130 et la frontaline 131. En comparaison avec les
énormes progrès réalisés dans la préparation d'alcools secondaires énantiomériquement purs, la synthèse d'alcools tertiaires demeure un sujet de
recherche actuel.
La méthode classique pour préparer ces alcools est la séparation de sels diastéréoisomériques.~~~ Une autre méthode fréquemment employée est la
transformation de produits naturels tels que les terpènes, les acides aminés, les

carbohydrates.. -148 L'utilisation de méthodes enzymatiques permis d'obtenir des résultats satisfaisants?g
90
a également
La synthèse par addition de réactifs organométalliques sur des cétones asymétriques est une approche prometteuse.lS0 La dihydroxylation d'oléfines 1 ,l disubstituées est de loin la méthode la plus efficace pour la préparation d'alcools tertiaires optiquement actifs.151 De même, I'époxydation asymétrique d'oléfines 1 ,l-disubstituées suivie du traitement avec une base ou un nucléophile conduit à ces alcools.152
La formation stéréocontrôlée de 1.3-diols est également d'une grande
importance pour les chimistes de synthèse puisque ce motif forme le squelette
de base de nombreux produits naturels biologiquement actifs tels que les macrolides. De nombreuses méthodes ont été développées pour la synthèse de ces diols avec contrôle de la stéréochimie relative des deux centres.
Récemment, Kishi et wllaborateurs~53 ont publié les premiers travaux sur la synthèse du altohyrtin A 132, dont la structure comporte de nombreux motifs 1,3-diol. Les auteurs présentent d'ailleurs une nouvelle approche à la synthèse de 1 ,Bdiols, dont un des centres est quaternaire.

Notre stratégie permet la synthèse stéreocontrôlee de 1'3-diols bis-
tertiaires. Le premier centre provient d'une époxydation asymétrique sur un alcène 1 '1-disubstitué, tandis que le deuxième centre résulte de la manipulation appropriée des substituants en a du carbonyle sur la
butyrolactone dérivée de l'époxyde 119. La voie de synthèse présentée conduit à un 1,3-diol syn. La stéréochimie des centres peut également être contrôlée de manière à obtenir un 1'3-diol anti . Cette approche est flexible et pourrait permettre l'obtention de synthons ayant des substituants variés.
4.4 Svnthèse du bicvcle et Dreeive de s t é r é o c m I .
Le diol 129 est finalement soumis aux conditions d'alkoxycarbonylation-
~ a c t o n i s a t i o n . ~ ~ ~ ~ ~ Le bicycle 133 est obtenu comme seul et unique produit,
avec un très bon rendement, sous forme d'un solide blanc. Les centres
quaternaires sur le diol ont pour effet de ralentir la réaction, sans toutefois l'empêcher. En effet, la réaction nécessite un minimum de 48 heures alors qu'il
ne fallait pas plus de 24 heures pour les diols 53 et 80.
PdCI2, CuCh NaOAc, AcOH
CO, 25 OC 60 hres
TBDPSO
La structure du bicycle 133 permet la différenciation en RMN H des
divers éléments de la molécule. Ceci permet d'effectuer plus facilement des
experiences NOE visant à déterminer la position relative des groupements les uns par rapport aux autres. À titre de comparaison, la synthèse du bicycle 136, dérivé du composé 124, a été réalisée selon une séquence non optimisée.
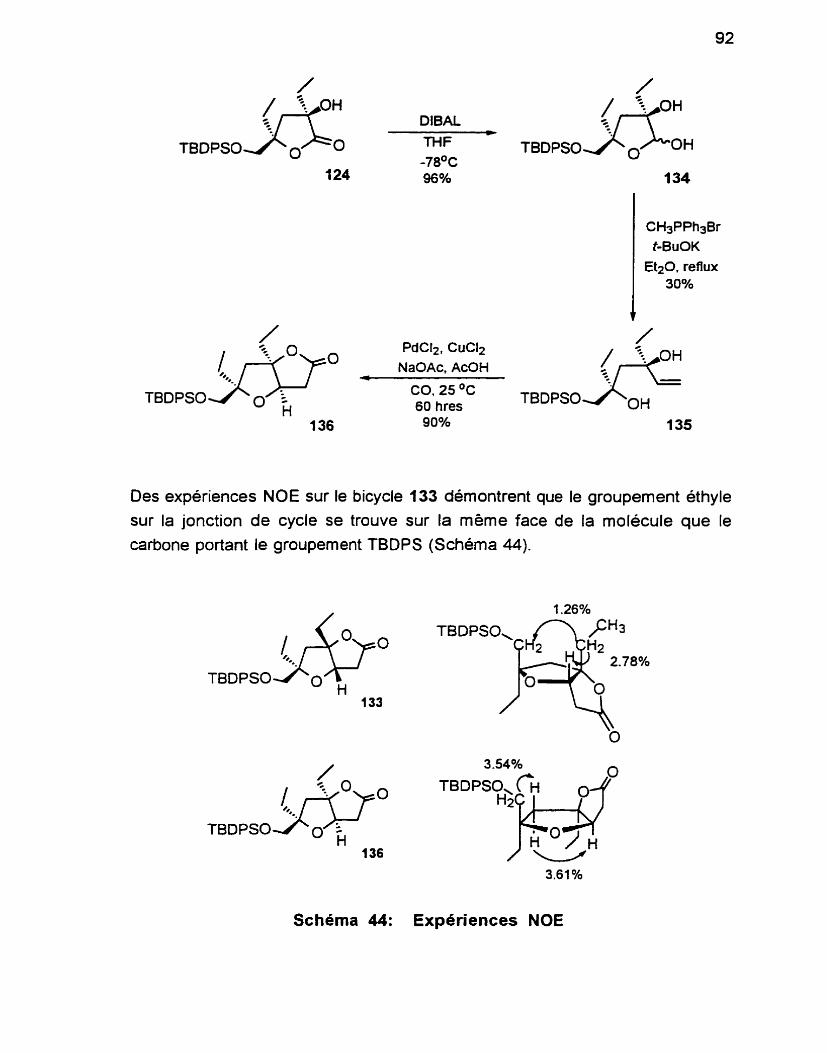
* TBDPSO THF
-78OC
/ PdC12, CuCl2
NaOAc, AcOH
TBDPSO CO, 25 OC 60 hres
TBDPSO OH
CH3PPh3Br t-BuOK
Et20. reflux 30%
Des expériences NOE sur le bicycle 133 démontrent que le groupement éthyle sur la jonction de cycle se trouve sur la même face de la molécule que le carbone portant le groupement TBDPS (Schéma 44).
TBDPSO
TBDPSO H
Schéma 44:
3.61 %
Expériences NOE

Les expériences NOE indiquent pour le bicycle 136 qu'il n'y a pas de rehaussement de signaux entre les protons du carbone portant le groupement
TBDPS et les groupements se trouvant sur la jonction de cycle. Ceci nous confirme que le composé 123 obtenu de la réaction d'hydroxylation possède le groupement OH anti par rapport au carbone portant le groupement TBDPS.
La stéréochimie du bicycle 133 a également été prouvée tout récemment par rayons-X (Schéma 45).
4.5 Voie a-ve de ~ r é n w t i o n du I.3-di01 s v ~
Une autre séquence de réactions a été tentée afin d'accéder plus
rapidement et plus efficacement au 1,3-diol syn. Le composé 122 est réduit avec DIBAL pour conduire au lactol 137, avec un bon rendement.
TBDPSO a DIBAL b
THF, - 78 'C TBDPSO OH
1 MsCI, Et3N CHzClz reflux 45 %
+ &f,"H TBDPSO
AD-Mix-p P
TBDPSO OH - 5 0 % TBDPSO
Le lactol 137 est ensuite déshydraté en générant le mésylate qui est ensuite éliminé pour conduire au composé 138 avec un médiocre rendement.154 L'alcène 138 est finalement soumis à une dihydroxylation avec le AD-mix-P
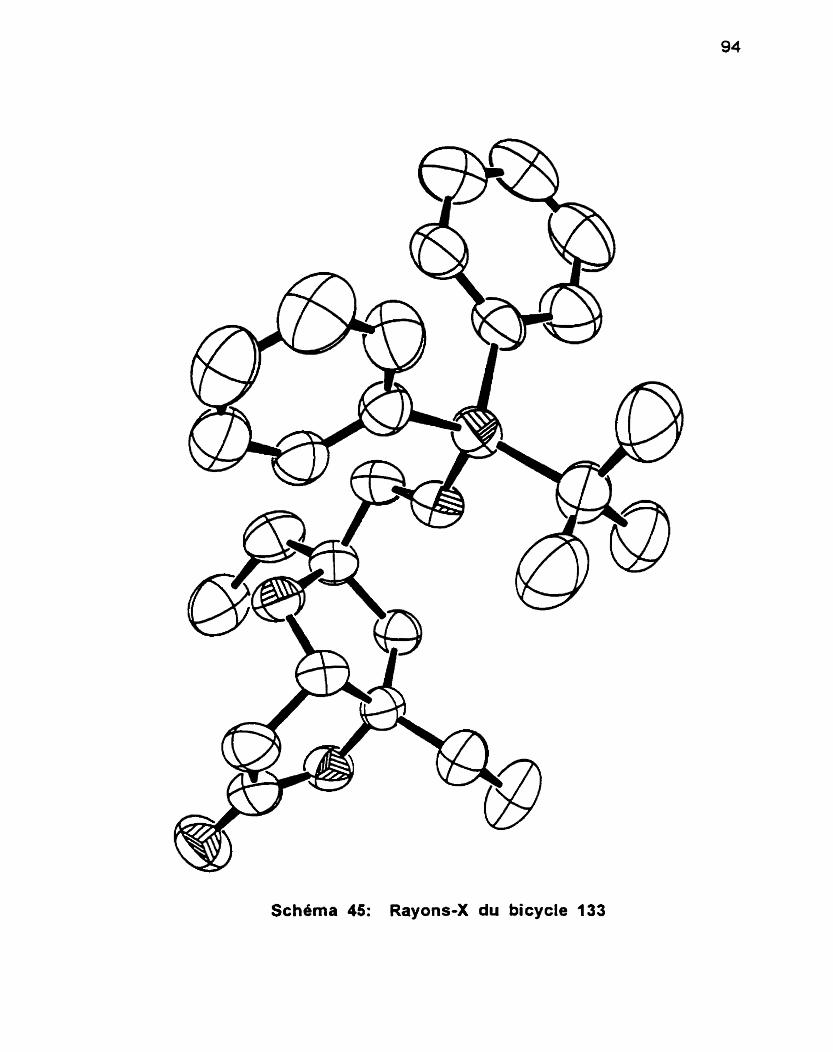
Sch4rna 45: Rayons-X du bicycle 133

(disponible commercialement) selon les conditions développées par Sharple~s.151~1~5 Le lactol 128 est obtenu majoritairement dans un rapport de 11:1, ce qui est en accord avec des travaux de Curranl56 qui utilise le modèle
proposé par Sharpless (Schéma 46).
I
AD-rn ix-a
Schéma 46: Selectivité de ia réaction de dihydroxylation
Les deux lactols peuvent être différenciés par RMN 1H et ils peuvent être séparés par chromatographie sur gel de silice. La différence majeure entre les deux composes (qui sont des mélanges de diastéréoisorneres) est donnée par le H porté par le C-2, soit
Cependant, le rendement de cette réaction est médiocre. Des essais pour
améliorer le rendement de la déshydratation et de la dihydroxylation se sont avérés infructueux. Cette voie a donc été abandonnée parce qu'elle s'avère
moins rentable que la précédente.
5 . Svnthase de la chaîne latérale
Tous ces travaux (ainsi que ceux de la section qui suit) ont été réalisés par Patrick Suter. en collaboration avec Yun-Xing Cheng, et les détails
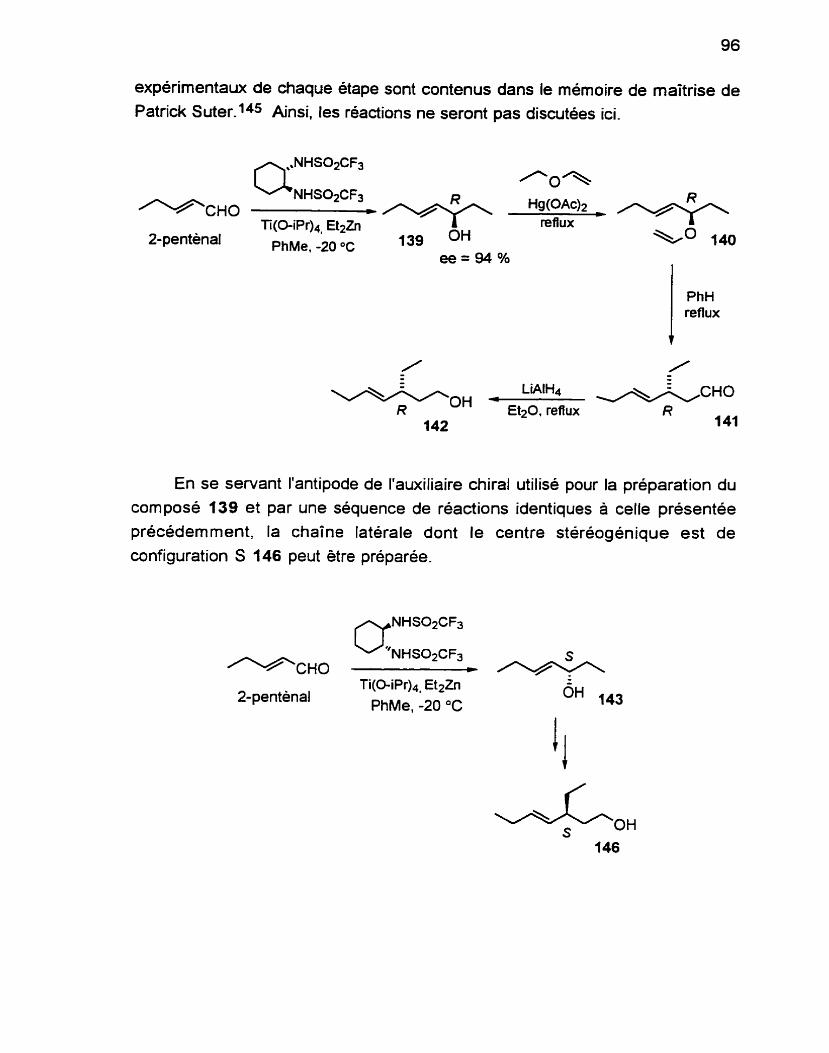
expérimentaux de chaque étape sont contenus dans le mémoire de maîtrise de Patrick S ~ t e r . ~ ~ ~ Ainsi, les réactions ne seront pas discutées ici.
-CHO
2- pentènal
NHS02CF3
QNHS02CF1 PO% R
Ti(O-iPr)4, Et2Zn PhMe, -20 OC 139 OH k0 140
ee = 94 %
PhH reflux
En se servant l'antipode de l'auxiliaire chiral utilisé pour la préparation du composé 139 et par une séquence de réactions identiques à celle présentée précédemment, la chaîne latérale dont le centre stéréogénique est de
configuration S 146 peut être préparée.

Les deux chaînes latérales ont été couplées avec l'unité bicyclique et en
toute fin, il s'avère que les données spectroscopiques et la rotation optique du produit final de synthèse contenant la chaîne dont le centre stéréogénique est de configuration R correspondent à celles du produit naturel. Les réactions suivantes ne seront donc décrites que pour la version de configuration R de la chaîne latérale. tout en gardant à l'esprit que la version S a été étudiée en parallèle.
Les étapes qui suivent permettent de transformer la chaîne en réactif de
Wlttig.
PPh3
PhH
Le groupement protecteur du bicycle 133 est clivé puis la fonction hydroxyle résultante est oxydée en aldéhyde selon les conditions de
Swerngglloo pour conduire à l'aldéhyde 152. L'ylure du composé 150 est
généré à l'aide de la base NaHMDS puis traité avec I'aldéhyde 152 pour conduire au composé 153 selon un couplage de type Wittig. La double liaison
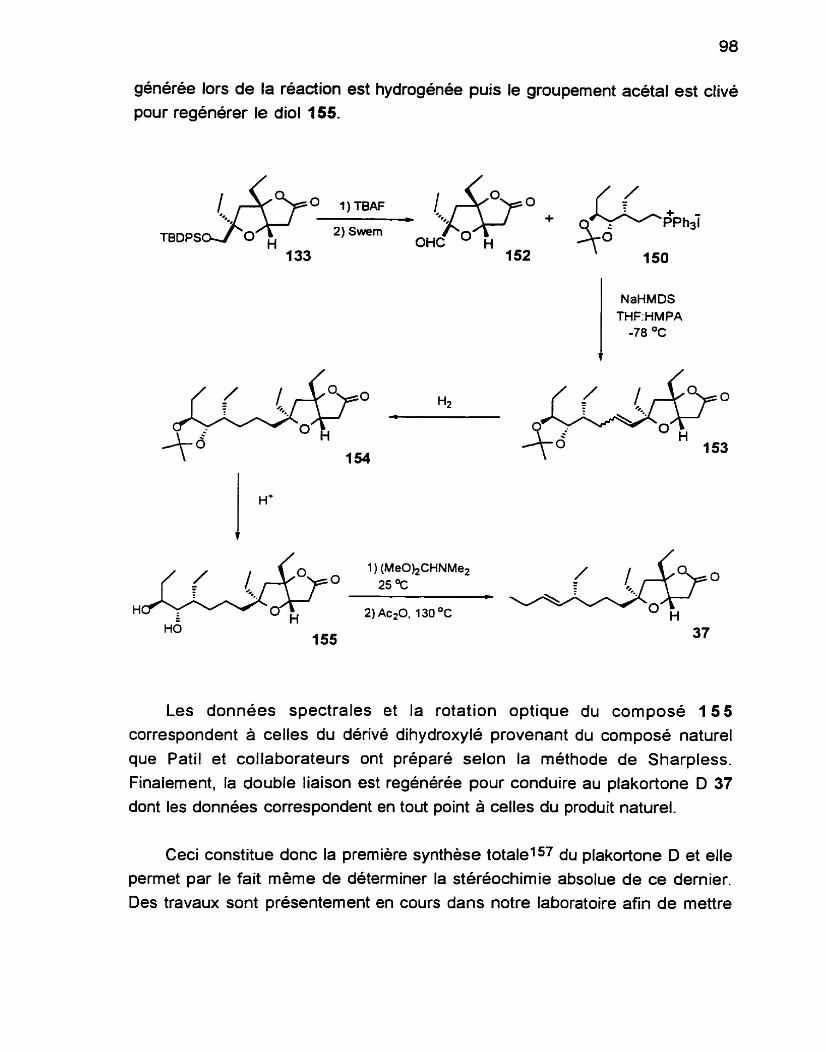
générée lors de la réaction est hydrogénée puis le groupement acétal est clivé
pour regénérer le diol 155.
NaHMDS THF:HMPA
-78 OC
Les données spectrales et la rotation optique du composé 1 5 5
correspondent à celles du dérivé dihydroxylé provenant du composé naturel que Patil et collaborateurs ont préparé selon la méthode de Sharpless.
Finalement, la double liaison est regénérée pour conduire au plakortone D 37 dont les données correspondent en tout point à celles du produit naturel.
Ceci constitue donc la première synthèse totale157 du plakortone D et elle
permet par le fait même de déterminer la stéréochimie absolue de ce dernier. Des travaux sont présentement en cours dans notre laboratoire afin de mettre

en application notre stratégie pour la synthèse des autres membres de la famille des plakortones.

CONCLUSION
Avant de clore ces travaux. il s'avère important de mettre l'emphase sur les résultats obtenus afin de mettre en &idence leur application à des projets futurs.
Nous avons présenté une nouvelle approche permettant l'accès à des tétrahydrofuranes 2.5-dialkyl-3-oxysu bstitues. Cette stratégie repose sur une réaction d'al koxycarbonylation-lactonisation catalysée par le Pd( Il). Notre approche constitue une utilisation inédite et innovatrice de cette réaction.
Cette stratégie est générale et très flexible puisque notre cheminement permet l'accès à deux familles de tetrahydrofuranes distinctes. Par une manipulation judicieuse, les tétrahydrofuranes (ZR. 3R. 5R) et (2S, 3s. 5R) peuvent être obtenus.
La synthèse énantiosélective du (-)- trans-kumausyne, isolé des algues rouges du genre Laurencia. a été réalisée. Celle-ci s'avère beaucoup plus concise et efficace que celles publiées à ce jour. II est envisageable d'appliquer la stratégie d6veloppée à la synthèse d'autres membres de cette famille.
Nous avons présenté également une approche à la synthese des divers tétrahydrofuranes isolés des algues brunes Notheia anomala. Tel que mentionné. ces travaux sont en voie d'être complet& dans notre laboratoire. Notre synthèse constituera donc une voie innovatrice à la préparation de ces compos6s souvent utilisés pour tester de nouvelles méthodologies de synthese.

Finalement, nous avons appliqué notre stratégie a la çynthése d'un nouveau composé naturel d'origine marine. le plakortone D. qui démontre des propriétés intéressantes pour le traitement de certaines maladies cardiaques. Nos travaux constituent la première synthèse de ce produit et par le fait même. permettent de prouver la stéréochimie absolue de ce dernier.
Lors de ces derniers travaux. nous avons réalisé la préparation d'un nouveau synthon chiral. soit un 1.3-diol syn optiquement actif dont les deux centres sont quaternaires. Notre approche est tout-à-fait inédite puisqu'elle donne lieu à un synthon possédant deux caractéristiques très recherchées en synthèse de produits naturels. En effet, le motif 1.3-diol se retrouve dans de nombreux composes. de même que les fonctions hydroxyles tertiaires. La stéréochimie des centres peut également être contrôlée de manière à obtenir un 1.3-di01 anti Cette approche est flexible et pourrait permettre l'obtention de synthons ayant des substituants variés. II est envisageable de croire que ces synthons pourraient trouver de nombreuses applications en synthese.


À moins de mention contraire, les réactions sensibles à l'humidité ont été effectuées dans de la vaisselle séchée à la flamme, sous atmosphère d'azote. Les liquides, solutions et solvents anhydres ont été transférés par seringue ou
via cannula à travers un septum.
Les spectres RMN'H et 13C ont été enregistrés sur un spectromètre Brüker
300 et les spectres IR sur un spectromètre Perkin-Elmer 781. Les points de fusion ont été mesurés dans des tubes capillaires ouverts à l'aide d'un appareil
de type électrothemal Thomas Hoover et ne sont pas corrigés. Les pouvoirs rotatoires ont été mesurés avec un polarimètre Jasco DIP-360.
Les chromatographies de type éclairis8 ont été réalisées sur gel de silice
(Kieselgel 60, 0.040-0.063 mm) de la compagnie EM Science. L'acétate d'éthyle, le dichlorométhane, I'hexane et l'éther de pétrole 35-60 O C étaient utilises après distillation. Les chromatographies sur couche mince ont été réalisées en utilisant les plaques de verre de la compagnie EM Science (0.25 mm d'épaisseur, F-254, no. 5714). De plus, les révélateurs employés étaient les suivants et ils étaient préparés selon les données indiquées:
219 de (NH4)6Mo7024.4H20 et 1 g de Ce(S04)2 dans 500 mL de 10% de H2S04 dans H z 0 1) 5% de H2S04 dans MeOH 2) 5% de vanilline dans MeOH
II est à noter que la référence en spectroscopie RMN I H et l3C est, pour tous les spectres, le signal du solvant utilisé et que le déplacement chimique

est donné selon l'échelle 6, en ppm. De plus, la convention suivante a été
adoptée afin d'alléger la description des spectres:
S = d = t =
9 = m = SI = dd = ddd = td =
singulet doublet
triplet quadruplet
multiplet singulet large doublet de doublets doublet de doublets de doublets
triplet dédoublé
Dans le cas de mélanges de deux diastéréoisomères, les signaux corres-
pondant à l'isomère minoritaire sont indiqués entre crochets.
En spectroscopie infra-rouge, la position des bandes est notée en cm-'.
De même, chaque valeur de rotation spécifique correspond à la moyenne de
20 lectures.
Les analyses élémentaires ont été réalisées a l'un ou l'autre des trois endroits suivants: Université de Genève (Labo de chimie pharmaceutique,
Service de microbiochimie, Suisse); Guelph Chernical Laboratories (Ontario);
Université de Montréal (Québec). Les spectres de masse haute résolution ont
été effectués au laboratoire de spectroscopie de masse de l'université de Sherbrooke (Québec). Les rayons-X ont été réalisés par le Professeur John
Bridson de l'université de Terre-Neuve.
Les solvants ou réactifs mentionnés ici ont été purifiés de la manière suivante:' 59
Éther: Hexane: THF: Toluène:
MeOH:
distillé sur sodium et benzophénone distillé sur sodium
distillé sur sodium et potassium distillé sur sodium distillé sur CaH2

C H2C 12: distillé sur CaH2 Et3N: distillée sur KOH P yridine: distillée sur KOH
CH3CN: distillé sur CaH2 1 ,3-diaminopropane: distillé sur CaH2
Le DMF anhydre a été acheté de la compagnie Aldrich.
Dans un tricol de 500 mL, on place l'acide D-glutamique (25.0 g, 0.17 mol) en suspension dans de I'eau (85 mL). On agite fortement la solution. On ajoute
simultanément une solution de NaN02 (17.9 g, 0.26 mol) dans de I'eau (25 mL) et une solution de HCI 5.6 N (42.5 mL, 0.24 mol) sur une période de 2.5 heures. On maintient le tricol dans un bain de glace tout au long de l'addition et pour encore 2.5 heures supplémentaires. On laisse ensuite agiter pour 15 heures à température ambiante. L'eau est enlevée par co-évaporation avec du benzène, en prenant soin de garder la température audessous de 50 O C . On ajoute ensuite 700 mL dlAcOEt et du Na2S04. Le mélange est agité 3 heures (agitation mécanique) puis filtré sur Büchner. Le résidu solide est lavé soigneusement avec de I'AcOEt. Le filtrat recueilli est ensuite agité avec de la
résine AG50WX2 pour 15 minutes, afin d'éliminer les acides aminés résiduels. On filtre le mélange et on évapore le solvant. On procède à une dernière CO-
évaporation avec du benzène. Le résidu est recristallisé dans un mélange

Ac0Et:benzène:éther de pétrole 35-60 O C pour conduire au composé 46
(13.95 g, 63%).
C5H604 Solide jaune pâle
130.10 g 1 mol
70-7 1 O C (AcOEt: benzène:éther de pétrole
3540 OC) litt.70: 73.5-74.0 O C
-14.7 (C 1-12, EtOH)
1itt.70: -1 5.9 (C 1.1 O, EtOH)
IR (KBr), vmax (cm-'): 3500-2500 (OH, acide), 1 800-1 700 (C=O, lactone et acide), 141 0 (CHz), 1230. 1 1 70
et 1060 (C-O).
RMN l H (DMSO), 6 (ppm):
RMN I3C (DMSO), 6 (ppm): 176.8, 171.4, 75.4, 26.7 et 25.4.
Les données sont en accord avec celles retrouvées dans la littérature?
A la solution du composé 46 (13.7 g, 105.0 mmol) dans du THF (65 mL), on ajoute, sur une période de 30 minutes, le complexe borane-sulfure de
diméthyle (2.0 M dans du THF, 56.0 mL. 112.0 rnmol), à O O C . Après l'addition,

on laisse agiter encore 30 minutes à O O C , puis 1.5 heures à température ambiante. On refroidit à nouveau la solution dans un bain de glace puis on ajoute lentement du MeOH (25 ml). Le solvant est évaporé puis on ajoute à nouveau du MeOH. La procédure d'évaporation est répétée 4 fois de manière à éliminer le triméthylborate formé. Le résidu obtenu est ensuite séché par CO-
évaporation (3 fois) avec du benzène. On recueille un liquide qui est purifié par chromatographie éclair en utilisant comme éluant un mélange de MeOH (2%) dans du CH2C12. On recueille 8.2 g (67%) d'un liquide jaune pâle.
C5H803
Liquide jaune pâle Rf
IR (film), v, (an-' ):
RMN 'H (CDC13), 6 (ppm):
116.12 g / mol
0.21 (2%MeOH 1 CHzC12)
3550-3200 (OH), 2930 (CH aliphatiques),
1780-1 720 (C=O, iactone), 1465, 141 0
(CHZ)~ 1350, 1 180 et 1060 (C-O).
4.54 (1 H, m l m-CH2-OH),
3.75 (1 Hl dd, J = 2.8, 12.6 HZ, -CH-ÇIIH-
OH), 3.54 (AH, dd, J ~ 4 . 5 , 12.6 HZ, -CH-çt[H-
OH), 3.12 (1 Hl SI OH),
2.54 (2H, m, CO-CH7-CH2), 2.18 (IH, m, -CO-CH2-UH) et 2.02 (1 H, m, -CO-CH2-ÇtLH).
RMN 13C (CDC13). 6 (ppm): 178.4, 81.2, 63.8, 28.7 et 23.1.
Les données sont en accord avec celles retrouvées dans la littérature.70

1.3 (5R)-4,5-dihydro-5-(t-butyldiph8nylsilyloxym~thyl)-2(3H)- furanone
La Et3N (14.2 mL, 101.9 mmol) est ajoutée à la solution du composé 47 (7.9 g, 68.0 mmol) dans du CH2C12 (70 mL), à O O C . On laisse agiter 10 minutes puis on additionne le DMAP (110 mg, 0.9 mmol) et le TBDPSCI (19.3 mL, 74.2 mmol). On laisse agiter encore une heure à O OC, puis 18 heures à température ambiante. Le mélange est lavé successivement avec des
solutions de HCI IO%, NaHC03 saturée puis NaCl saturée. La phase
organique est ensuite séchée sur MgS04, filtrée puis concentrée. Le résidu obtenu est recristallisé dans de I'hexane pour conduire au composé 48 (20.0 g,
83%).
C21 Hî603Si Solide blanc
RMN 1H (CDC13), 6 (ppm):
354.52 g 1 mol
73-75oC (hexanes) litt.77: 72-73 OC
-33.7 (C 1-15, EtOH),
-27.8 (C 2.08, CHC13) litt.77: -22.6 (c 1.00, CHCI3)
3040 (C-H aromatiques), 2940, 2850 (C-H aliphatiques), 1765 (C=O, lactone), 1420,
1460 (CHz), 1170 et 1100 (C-O).
7.67 (4H, m, H aromatiques),
7.36 (6H, m. H aromatiques), 4.60 (1 H, m, CH-CH2-OTBDPS), 3.89 (IH, dd, J = 3.4 HZ, 11.4 HZ, CH- CHH-OTBDPS),

RMN l3C (CDC13), 6 (ppm):
3.69(1H, dd, J = 3.4 HZ, 11.4 HZ, CH- S;tLH-OTBDPS), 2.68 (1 HI m, CH2-mH-CH), 2.51 (AH, m. CH2-ÇtLH-CH), 2.27 (2H. m, Cl+-CHz-CH) et
1 -06 (9H, s, t-butyle).
Les données sont en accord avec celles retrouvées dans la littérature.77
1.4 Hydroxylation de la butyrolactone 48
À la solution de LiHMDS (1 .O M dans du THF, 10.5 mL, 10.50 mmol), à -78
OC, on ajoute lentement le composé 48 (1.50 g, 4.23 mmol) dans du THF (60
mL). On laisse agiter une heure à -78 O C puis on ajoute le MoOPH (2.80 g, 6.45 mmol). La solution est ensuite agitée pour 3 heures additionnelles à cette
température. On ajoute alors un peu d'une solution de Na2S03 saturée et de l'éther puis on laisse le mélange revenir à température ambiante. On agite
fortement la solution et ajoute suffisamment d'eau pour dissoudre tous les sels.
On évapore la majeure partie du THF puis on extrait ensuite le résidu plusieurs fois à l'éther. Les phases organiques réunies sont lavées avec une solution de HCI IO%, puis avec NaHC03 saturée puis enfin une solution de NaCl saturée. Après séchage des phases organiques sur MgS04, filtration et concentration, le résidu est purifié par chromatographie éclair avec comme éluant un mélange

Et2O:éther de pétrole 35-60 OC:C H Cl3 (45:50:5) pour conduire majoritairement au composé 49 (1.00 g, 64%). de même qu'une petite quantité de son epimère
en C-2 50 (143 mg, 9.1 %). Le composé 49 est ensuite recristaliisé dans de
I'hexane.
370.52 g I mol
0.21 (Et0:éther de pétrole 3540 O C :CHCI3 4550:s)
P-f.: 81 8 2 O C (hexanes)
IR (KBr), vmax (cm-'): 3540-3320 (OH), 3060, 3040 (CH aroma-
tiques), 2950, 2920, 2850 (CH aliphati-
ques), 1770 (C=O, lactone), 1465, 1420
(CHz), 1 180.1 100 et 1070 (C-O).
RMN 1H (CDC13), 6 (ppm): 7.64 (4H, m, H aromatiques),
7.43 (6H, ml H aromatiques), 4.81 (1 Hl t, J = 8.8 HZ, çtl-OH), 4.66 (1 H, m, ÇtL-CH2-OTBDPS),
3.90 (1H, dd, J ~ 2 . 6 Hz, 11.5 Hz, CH-
UH-OTBDPS), 3.63 (lH, dd, J = 2.6 HZ, 11 -5 Hz, CH- ÇtLH-OTBDPS),
3.35 (IH, SI, OH), 2.63 (1 Hl m, (0H)CH-ÇtlH-HCO-),
2.36 (IH, rn, (0H)CH-ÇtLH-HCO-) et i -05 (9H, s, t-butyle).
RMN 13C (CDCIj), 6 (ppm): 178.3, 135.6, 132.6, 130.1, 128.0, 77.7,
67.6, 65.3, 32.8, 26.8 et 19.2.
Analyse élémentaire: calculée: H 7.07 C 68.07;
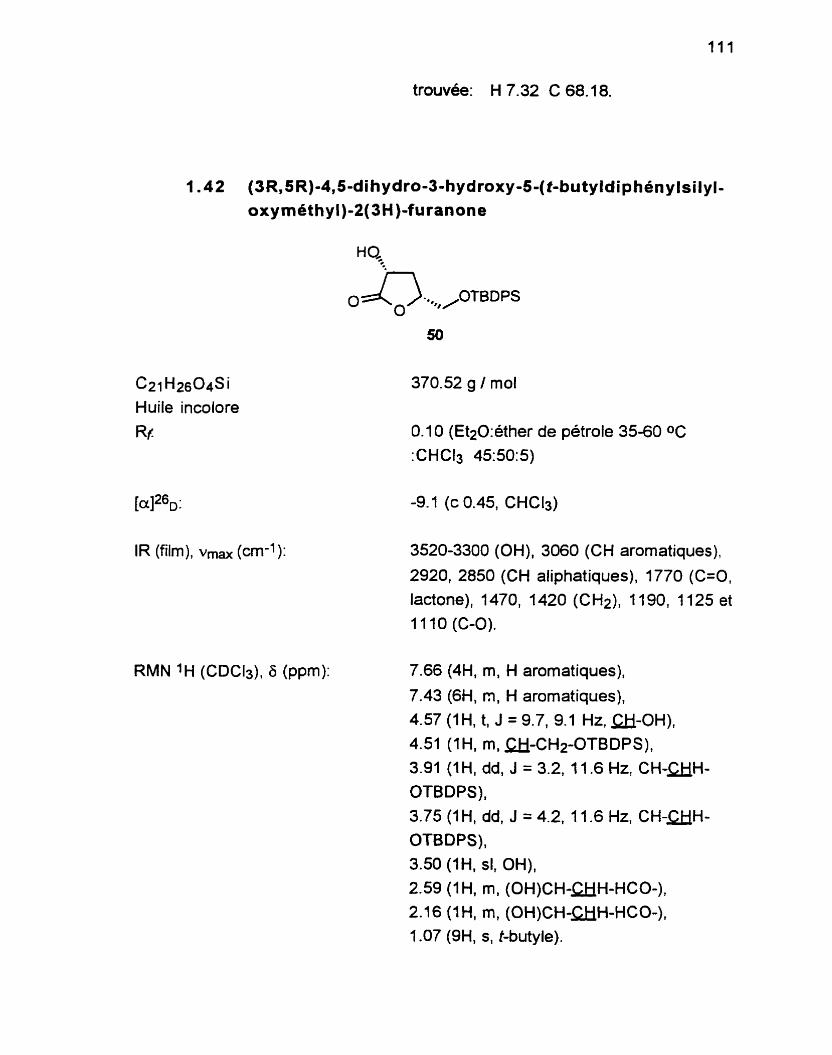
trouvée: H 7.32 C 68.18.
C21 H2604Si
Huile incolore
Rt:
IR (film), VM, (cm-' ):
RMN AH (CDC13). 6 (ppm):
370.52 g 1 mol
0.1 0 (Et2O:éther de pétrole 35-60 OC :CHCI3 45:50:5)
3520-3300 (OH), 3060 (CH aromatiques),
2920, 2850 (CH aliphatiques), 1770 (C=O, lactone), 1470, 1420 (CHz), 1 190, 1 125 et Il 10 (C-O).
7.66 (4H, m, H aromatiques),
7.43 (64 m. H aromatiques), 4.57 (1 H, t, J = 9.7, 9.1 HZ, =-OH), 4.51 (1 H. m. ÇtL-CH2-OTBDPS), 3.91 (IH, dd, J = 3.2, 11.6 HZ, CH-çtLH- OTBDPS), 3.75(1HI dd, Jz4.2 , 11.6Hi, CH-UH- OTBDPS), 3.50 (IH, SI, OH), 2.59 (1 H, m, (0H)CH-ÇtLH-HCO-), 2.1 6 (1 H, m, (0H)CH-ÇtlH-HCO-), 1 .O7 (9H, s, t-butyle).

RMN 1% (CDC13), 6 (ppm): 177.3, 135.7, 132.8, 130.0, 127.9, 77.2,
68.3, 64.5, 32.4, 26.8 et 19.3.
Masse exacte mle: (ICI ammoniaque)
SM (IE, 70 Ev)
pour C21 H30N04Si (MNH4+), calculée: 388.1 944; trouvée: 388.1 940.
A la solution du composé 50 (50 mg, 0.135 mmol), de la PPh3 (54 mg,
0.206 mmol) et de l'acide benzoïque (33 mg, 0.270 mmol) dans de l'éther (5 mL), on ajoute le DEAD (32 PL, 0.203 mmol) goutte à goutte, à -20 O C . Le
mélange est agite 3 heures à cette température. Le solvant est ensuite évaporé et le résidu est dilué dans une solution de Ac0Et:Hexanes (1 5:85). Le solide qui se forme est filtré et la solution est de nouveau évaporée. Le résidu est purifié par chromatographie éclair en utilisant un mélange Et2O:éther de pétrole 35-60 O C (25:75) pour conduire au composé 51 (60 mg, 94%).
C28H3005Si
Huile incolore
Rf:
474.63 g 1 mol
0.20 (Et20:éther de pétrole 35-60 OC 1 :3)

IR (film). v,, (cm-1):
RMNlH (CDCI3), 6 (pprn):
RMNI3C (CDC13), 6 (pprn):
Masse exacte m/e: (IC, isobutane)
3060 (CH aromatiques), 2945, 2845 (CH
aliphatiques), 1790 (C=O. lactone), 1725 (C=O, ester). 1600, 1 580. 1470, 1450, 1425 (CHz), 1265, 1 180 et 1 1 15 (C-O).
8.08 (3H, ml H aromatiques de l'ester),
7.70 (4H, rn, H aromatiques du TBDPS),
7.62 (2H, m, H aromatiques de l'ester), 7.43 (6H, m, H aromatiques du TBDPS), 5.76 (1 H, t, J = 9.4 HZ, PhCOO-ÇH-CO-), 4.62 (1 HI m. O-CH-CH2-OTBDPS), 3.97 (IH, dd, J = 3.5, 11.7 HZ, -CHH- OTBDPS), 3.80 (1 HI dd, J= 4.0. 1 1 -7 HZ, -CH& OTBDPS), 2.83 (1 HI m, -çHH-CH-CH2-OTBDPS), 2.41 (1 H, m, -çtLHCH-CH2-OTBDPS) et 1.08 (9H, s, t-butyle).
pour C23H31 O5Si (MH+), calculée: 475.1941;
trouvée: 475.1931.
SM (IE, 70 eV) 41 7 (M - t-butyle, 20), 1 05 (1 00).

A la solution du composé 51 (26.1 mg, 0.055 mmol) dans du méthanol (2 mL), on ajoute du K2C03 (16.0 mg, 0.1 16 mmol). On laisse agiter quelques
heures à température ambiante puis on évapore le solvant. Le résidu est alors
dissout dans de I'AcOEt et on y ajoute un peu d'une solution de HCI 10%. On laisse le mélange agiter vigoureusement pour au moins deux heures. Les deux
phases sont ensuite décantées et on lave la phase organique plusieurs fois
avec une solution saturée de NaHC03 puis avec une solution saturée de NaCI.
Après séchage de la phase organique sur MgS04, filtration et évaporation, le résidu est purifié par chromatographie éclair en utilisant un mélange Ac0Et:éther de pétrole 35-60 O C (3:7). On recueille 16.1 mg (79%) du composé 50 dont les caractéristiques ont été énumérées auparavant.
Le complexe borane-tétrahydrofurane (1.0 M dans THF, 24.3 mL. 24.30
mmol) est ajouté à la solution du composé 50 (4.51 g, 12.17 mmol) dans du THF (360 mL), à O OC, sur une période de 30 minutes. On laisse alors agiter à température ambiante pour toute la nuit. On ajoute ensuite un peu d'eau, jusqu'à ce qu'il cesse d'y avoir dégagement de H2. On effectue plusieurs co-

évaporations avec une solution de 2% dlAcOH dans du MeOH, puis avec du
benzène afin d'éliminer l'eau. Le résidu est purifié par chromatographie éclair en utilisant comme éluant un mélange AcOEt:CH2C12 (2:8) pour conduire au lactol 52 (2.95 g, 65%), sous forme d'un mélange d'isomères.
C2lH2804Si Huile incolore
Rfi
372.54 g 1 mol
0.20 (AcOE~:CH~CIZ 2:8)
IR (film), vmax ( c d ) : 3500-3240 (OH), 3060 (CH aromatiques),
2920, 2845 (CH aliphatiques), 1425 (CHz), 1 105 et 1030 (C-O).
RMN 1H (CDC$), 6 (ppm): 7.71(4H1 ml H aromatiques),
7.43 (6H, m, H aromatiques),
5.42 [5.18] (1 HI SI [dd,J = 3.3 Hz, 9.1 Hz],
(HO)-çtl-O-), 4.39 (1 Hl ml CH2-m-OH),
4.1 5 (2H, ml -O-ÇtL-CH2 et OH), 3.84 (1 Hl m. CH-ÇtLH-OTBDPS). 3.48 (1 H, m, CH-ÇtLH-OTBDPS),
2.54 [2.33] (1 HI rn, (0H)CH-UH-HCO),
1.98 (2H, ml (OH)CH-;tlH-HCO- et OH) et
1.10 [1.08] (9H, S. f-butyle).
RMN 13C (CDC13), 6 (ppm):
Analyse élémentaire: calculée: H 7.58 C 67.71; trouvée: H 7.80 C 67.87.

1 .8 (1 R, 3R)-l,3-dihydroxy-l-(t-butyldiphBnylsilyloxyméthyl)-4- pentène
À la suspension de t-BuOK (200 mg, 1.68 mmol) dans de l'éther (65 mL), on ajoute le CH3PPh3Br sec (600 mg, 1.68 mrnol), à température ambiante. On laisse agiter 2 heures puis on ajoute le lactol 52 (250 mg, 0.67 mmol) en solution dans de l'éther (10 mL). Le mélange est agité toute la nuit à température ambiante. On ajoute alors un peu d'eau et on extrait plusieurs fois avec un mélange Et20:éther de pétrole 3540 O C (21). Les phases organiques réunies sont séchées sur Na2S04, filtrées puis concentrées. Le résidu est
purifié par chromatographie éclair en utilisant comme éluant un mélange Et2O:éther de pétrole 35-60 O C (7:3) pour conduire au diol 53 (160 mg, 64%).
C22H3003Si
Huile incolore
Rt
RMN 1H (CDCIB), 6 (ppm):
370.56 g / mol
0.30 (Et2O:éther de pétrole 35-60 O C 7:3)
4.3 (c 1.4, EtOH)
-5.6 (C 1.2. CHC13)
3500-3260 (OH), 3060 (CH aromatiques),
2940, 2930 (CH aliphatiques), 1420 (CHî), 11 10 et 1070 (C-O).
7.68 (4H, m. H aromatiques),
7.40 (6H. m. H aromatiques), 5.87 (1 H, m. H2C=m-CH(OH)), 5.28 (lH, d, J = 18.8 Hz, BtiC=CH-
W O H ) ) , 5.12 (IH, d, J = 10.3 HZ, HHCzCH-
W W ) ,

4.43 (1 Hl ml H2C=CH-Çtl(OH)), 4.06 (1 H, m, (OH)çt[-CHTOTBDPS), 3.67 (1 H, dd, J = 4.2 HZ, 10.2 HZ, CH- CHH-OTBDPS), 3.58 (1 H, dd, J = 7.3 HZ, 10.2 HZ, CH-
CHH-OTBDPS), 2.66 (1 H, SI, OH), 2.20 (1 Hl si, OH), 1.72 (1 H, m. (0H)CH-WH-CH(OH)), 1.51 (1 Hl m, (0H)CH-S;LLH-CH(0H)) et 1.08 (9H. s, t-butyle).
RMN 13C (CDC13). 6 (ppm): 140.7, f 35.4, 133.0, 129.7, 127.7, 114.1,
69.9, 69.3, 67.8, 38.6, 26.8 et 19.1.
Masse exacte mie: (IC, ammoniaque)
pour C22H34N03Si (MNH4*), calculée: 388.2308; trouvée: 388.2302.
SM (IE, 70 eV) 295 (S), 241 (IOO), 199 (30).
*#t 0' OTBDPS
On place dans un tricol le PdCI2 (60% Pd, 3.9 mg, 0.022 mmol), le CuCI2 (1 10 mg, 0.648 rnmol) et le AcONa (71 mg, 0.864 mmol). On y ajoute ensuite la solution du di01 53 (80 mg, 0.216 mmol) dans de I'AcOH (15 mL). On fait
barbotter du CO dans la solution, qui passe initialement d'une couleur verte à

une couleur rouille. On laisse le mélange agiter ainsi pour 24 heures. Suite à cette période, le mélange est dilué dans du benzène puis filtré sur cellulose. La phase organique recueillie est lavée successivement avec des solutions de
NaHC03 saturée et NaCl saturée. Une huile est obtenue après séchage de la
phase organique sur MgS04, filtration et évaporation. Ce résidu est ensuite chromatographié selon la technique éclair en utilisant comme éluant un
mélange Et2O:éther de pétrole 35-60 OC (6:4) pour conduire au bicycle 58 (80
mg, 93%).
C23H2804Si Huile incolore
Rt:
396.56 g 1 mol
0.28 (Et2O:éther de pétrole 35-60 O C 6:4)
RMN 1~ (CDC13). 6 (pprn):
RMN 1% (CDC13), 6 (pprn):
Masse exacte rnle: (IC, ammoniaque)
3040, 3060 (CH aromatiques), 2945, 2920.
2845 (CH aliphatiques), 1780 (C=O, lac- tone), 1465, 1420 (CHz), 1180, 1140, 1105
et 1070 (C-O).
7.66 (4H, rn, H aromatiques),
7.40 (6H, m, H aromatiques),
5.00 (1 H, m, -CO-O-Çti-CH2-), 4.57 (1 H, m. -CO-CH2-çtl-O-),
4.13 (1 H, m. -O-ÇtL-CH2-OTBDPS), 3.72 (2H. ml -O-CH-w-OTBDPS),
2.70 (2H, ml CO-CH?-CH-O-),
2.36 (1 Hl m, -mH-CH-CH2-OTBDPS),
2.21 (1 H, rn, -mH-CH-CH2-OTBDPS) et
1 -06 (9H, s, t-butyle).
pour C23H32N04Si (MNH4+), calculée: 414.2100;

SM (IE, 70 eV)
trouvée: 41 4.2097.
339 (M - t-butyle, 1 OO), 199 (37), 105 (70).
- OTBDPS "9/
À une solution du bicycle 58 (80 mg, 0.20 mmol) dans du THF (4 m l ) a -78 OC, on ajoute lentement le DIBAL (1 .O M dans du toluène, 300 pl , 0.30 mmol).
On laisse agiter à cette température pour 3 heures puis on ajoute un peu d'une
solution de NH&I saturée. On laisse le mélange revenir à température
ambiante. Par la suite, on ajoute un peu d'une solution de HCI 10% et de
l'éther et la solution est agitée fortement pendant quelques minutes. On effectue plusieurs extractions a l'éther et les phases organiques réunies sont
lavées avec une solution saturée de NaHC03 puis avec une solution de NaCl saturée. Après séchage des phases organiques sur Na2S04, filtration et concentration, le résidu est purifié par chromatographie éclair en utilisant
comme éluant un mélange AcOEt:CH2C12 (5:95) pour conduire au lactol 59 (80
mg, 100%), sous forme d'un mélange d'isomères.
C23H3004Si
Huile incolore
RF
398.57 g 1 mol
0.20 (AcOEt:CH2C12 5:95)

IR (film), v,, (cm-'):
RMN 1H (CDCb), 6 (ppm):
3400 (OH), 3060, 3040 (CH aromatiques),
2920, 2950 (CH aliphatiques), 1465, 1420
(CH2). 1 100 (C-O).
7.68 (4H, m, H aromatiques),
7.40 (6H, m, H aromatiques), 5.55 [5.40] (IH, m, -çY(OH)), 4.81 [4.74] (1 Hl ml -CH(OH)-CH2-m-), 4.60 [4.50] (1 Hl m, -CH(OH)-O-=-), 4.15 [4.00] (1 H, m. -O-ÇY-CH2-OTBDPS), 3.97-3.56 (2H, ml -O-CH-CHPOTBDPS), 3.30 (1 Hl SI, OH), 2.51-2.16 (ZH, ml (0H)CH-CH2-CH-), 2.02 (2H, ml CH?-CH-CH2-OTBDPS) et 1 -07 [ A .OS] (9H, s, 1-butyle).
RMN 1% (CD&), 6 (ppm): 135.5 [135.4], 133.5 [132.9]. 129.5 [129.4],
127.6 [127.4], 100.0 [99.9], 84.6 [82.8], 83.5 [83.4], 81 -3 [80.7], 66.2 [64.0], 41.4
[40.2], 36.5 [34.9], 26.6 [26.6], 20.8 [19.1].
Masse exacte mie: (ICI ammoniaque)
SM (IE, 70 eV)
pour C23H34N04Si (MNH4+), calculée: 31 6.2257;
trouvée: 41 4.2250.
341 (M - t-butyle, 12), 263 (38), 199 (1 00).

1.1 1 Otéfination du lactol 59
1.1 1 1 (2R,3R,SR)-3-hydroxy-5-(t-butyldiph6nylsilyloxy- méthyl)-2-(2(E)-5-trimdthylsilyl-2-pentèn-4-ynyl)- tdtrahydrofurane
À la suspension de t-BuOK (191 mg, 1.62 mmol) dans de l'éther (75 mL), on ajoute le bromure de (3-triméthylsilyl-2-propynyl)-triphénylphosphonium sec (725 mg, 1.60 mmol), a température ambiante. On laisse agiter 3 heures puis on ajoute le lactol 59 (320 mg, 0.80 mmol) en solution dans de l'éther (25 mL).
Le mélange est agité toute la nuit à température ambiante. On ajoute alors un peu d'eau et on extrait plusieurs fois avec un mélange Et20:éther de pétrole 35- 60 O C (2:l). Les phases organiques réunies sont séchées sur Na2S04, filtrées puis concentrées. Le résidu est purifié par chromatographie éclair en utilisant un mélange Et20:éther de pétrole 35-60 oC:CHCI3 (10:85:5) pour conduire au composé 60 (335 mgt 85%) et son isomère cis 61 (31.5 mg, 8%).
C29H4003Si2
Huile incolore
RF
RMN 1H (CDC13), 6 (ppm):
492.81 g 1 mol
0.13 (Et2O:éther de pétrole 35-60 OC :CHCI3 10:85:5)
3430 (OH), 3060, 3040 (CH aromatiques),
2950, 2850 (CH aliphatiques), 2160, 2120
(CEC ), 1465, 1425 (CHz), 1245 (Si-CH3), 1 175, 11 05, 1070 (C-O).
7.68 (4H, m, H aromatiques),
7.40 (6H, m. H aromatiques),

RMN 13C (CDC13), 6 (ppm):
Masse exacte mle: (ICI ammoniaque)
SM (IE, 70 eV)
6.26(1H, td, J=15.9,7.4Hz1 C H = m -
CH2-1. 5.66 (IH, dl J = 15.9 HZ, -m=CH-CH2-),
4-21 (1 H, m. -=(OH)-CH2-), 4.08 (1 H, m. -Ou-CH2-OTBDPS), 3.87 (IH, d, J = 10.8 HZ, -CflH-OTBDPS), 3.75 (1 H, m. -O-ÇtL-CH2-CH=), 3.61 (1 H, d, J = 10.8 HZ. CHH-OTBDPS), 2.60 (2H, m, CH=CH-CH?-), 2.42 (1 H, m, CH(0H)-ÇtlH-), 2.06 (1 H, m, CH(0H)- - i f - ) , 1.61 (AH, SI, OH), 1 .O5 (9H, s, t-butyle) et 0.19 (9H. s, TMS).
pour C2gH44N03Si2 (MNH4+), calculée: 51 0.2860; trouvée: 51 0.2855.
435 (M - t-butyle, 18), 241 (48), 199 (1 00).
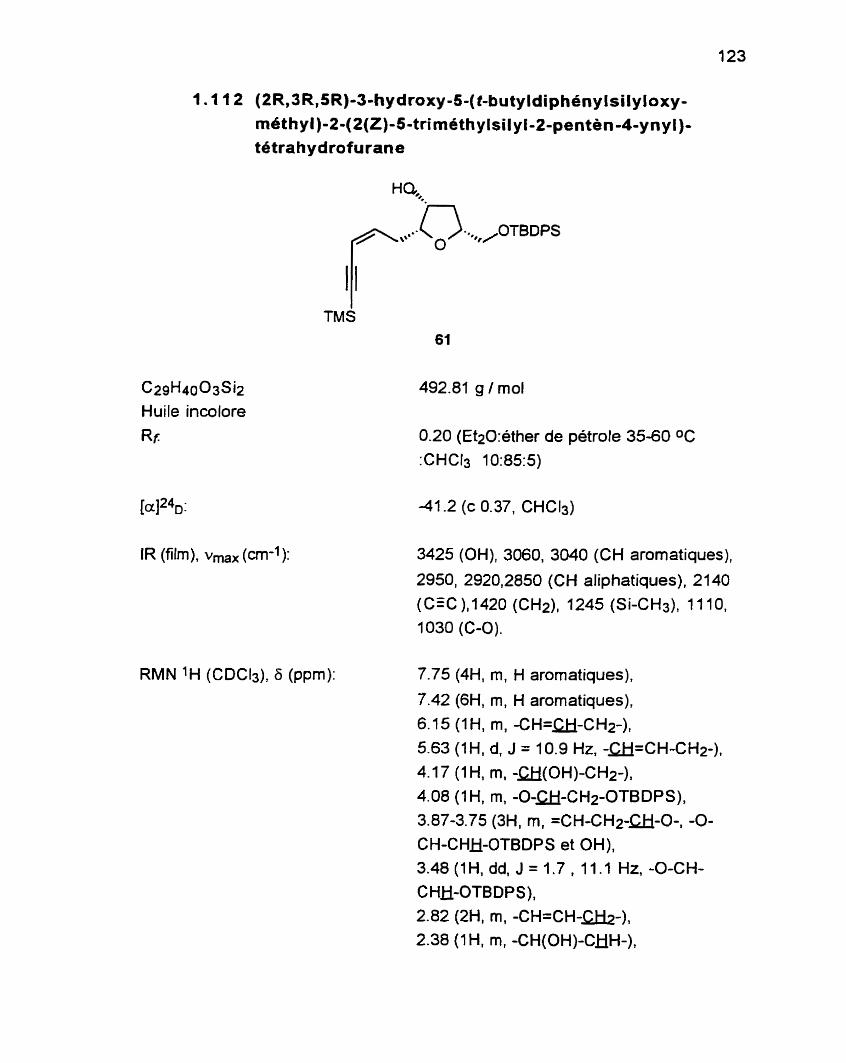
1.1 12 (2R,3R,SR)-3-hydroxy-5-(t-butyldiphénylsilyloxy- méthyl)-2-(2(Z)-5-trim~thylsilyl-2-penten-4-ynyl)- tetrahydrofurane
C29H4003Si2 Huile incolore
h
IR (film), v,, ( c d ):
RMN 1H (CDC13), 6 (ppm):
0.20 (Et2O:éther de pétrole 35-60 O C
:CHCI3 10:85:5)
3425 (OH), 3060, 3040 (CH aromatiques),
2950, 2920,2850 (CH aliphatiques), 2140
(CEC ),1420 (CHz), 1245 (Si-CH3), 11 10, IO30 (GO).
7.75 (4H1 m. H aromatiques),
7.42 (6H1 m. H aromatiques), 6.1 5 (1 HI ml -CH=m-CH2-),
5.63 (1 Hl d, J = 10.9 HZ, -CH=CH-CH2-), 4.17 (1 H, m, -çtl(OH)-CH2-), 4.08 (1 H, ml -O-u-CH2-OTBDPS), 3.87-3.75 (3H, ml =CH-CH2-u-O-, -O- CH-CHH-OTBDPS et OH), 3.48 (1 H, dd, J = 1.7 , 1 1.1 HZ, -O-CH- CHH-OTBDPS), 2.82 (2H, m, -CH=CH-CH7-), 2.38 (1 H, m, -CH(OH)-CHH-),

RMN 13C (CDC13), 6 (ppm):
2.08 (1 H, m, -CH(OH)-CHH-),
1 -09 (9H, s, t-butyle) et
0.18 (9H. s, TMS).
À la solution du composé 60 (216 mg, 0.44 mmol) dans de la pyridine (10 mL), on ajoute le DMAP (6.0 mg, 0.05 mmol) et l'anhydride acétique (145wL,
1.54 mmol). On laisse agiter pour deux heures à température ambiante. Si la
réaction ne semble pas complète après ce temps, on peut ajouter quelques
gouttes d'anhydride acétique. Le mélange est ensuite dilué avec de l'éther et
la phase organique est lavée successivement avec des solutions de HCI I O % ,
NaHC03 saturée et NaCl saturée. La phase organique est séchée sur MgS04, filtrée puis évaporée. Le résidu est purifié par chromatographie éclair en utilisant un mélange Et20:éther de pétrole 35-60 O C (2:8) pour conduire au
composé 62 (235 mg, 100%).
534.84 g 1 mol
0.52 (Et20:éther de pétrole 35-60 OC 2:8)
2.6 (c 1.02, CHC13)

IR (film), v,, (cm-'):
RMN 1H (CDC13), 6 (ppm):
3060, 3040 (CH aromatiques), 2950, 2920,
2850 (CH aliphatiques), 21 60, 21 40 ( C X ), 1735 (C=O, ester), 1465, 1420
(CHz), 1240 (Si-CH3), 11 1 O (C-O).
7.66 (4H, ml H aromatiques),
7.40 (6H, ml H aromatiques), 6.20 (1 H, m, -CH=m-CH2-),
5.60 (1 H,d, J = 15.9 HZ, -M=CH-CH2-), 5.33 (1 H, ml -çtl-OAc), 4.1 0 (1 HI m, -O-WH2-OTBDPS), 3.85 (1 Hl m, =CH-CH2-ÇH-O-) 3.77 (1 Hl dd, J = 3.8, 10.8 HZ, -0-CH-
CHH-OTBDPS), 3.61 (1 Hl dl J = 3.8, 10.8 HZ, -O-CH- CHH-OTBDPS),
2.41 (3H, m. -CH=CH-CH9- et CH(0Ac)-
ÇHW 1
2.09 (3H, s, OAc), 2.01 (1 H, ml CH(0Ac)-WH-),
1 .O5 (9H, s, t-butyle) et
0.19 (9H, S. TMS).
RMN 13C (CDC13), 6 (ppm): 170.5, 141.4, 135.6, 133.4, 129.7, 127.7,
112.0, 103.8, 93.5, 80.3, 77.8, 75.7, 66.0,
34.7, 33.4, 26.9, 21 -1, 19.2 et 0.5.
Masse exacte mie: (IC, ammoniaque)
pour C 31 H4304Si2 (Mi++). calculée: 535.2700; trouvée: 535.2694.
SM (IE, 70 eV) 477 (M - t-butyle, 40), 241 (1 OO), 1 63 (28).

A la solution du composé 62 (39 mg, 0.073 mmol) dans du CH3CN (3
mL), on ajoute l'hydrate du fluorure de benzyltriméthylamrnonium (40 mg) en
une seule portion, à O O C . On laisse ensuite revenir à température ambiante et la réaction est suivie par couche mince jusqu'à ce que tout le substrat soit
consommé. On dilue le mélange dans de I'AcOEt (10 mL) puis on filtre sur
Florisil. Après évaporation, le résidu est purifié par chromatographie éclair e n utilisant un mélange Ac0Et:éther de pétrole 35-60 OC (1:l) pour conduire au
composé 63 (15.5 mg, 95%).
C12H1604 Huile incolore
R 6
224.26 g / mol
0.22 (Ac0Et:éther de pétrole 3540 OC
1: l )
IR (film), v,, ( c d ): 3400 (OH), 3240 ('CH ), 2900,2830 (CH
aliphatiques), 1720 (C=O, ester), 1420
(CH2), 1230 et 1 1 10 (C-0).
RMN 1H (CDCI3), 6 (ppm): 6.20 (IH, td, J = 7.4, 15.9 Hz, -CH==-
CH 2-1, 5.53 (1 H, d, J = 15.9 HZ, -çH=CH-CH2-),
5.32 (1 H. m, -çH-OAc), 4.28 (1 H, rn, =CH-CH2-çHO-), 4.03 (1 H, m, -O-ÇY-CH2-OTBDPS
3.73 (1 Hl dd, J = 3.1, 1 1.9 HZ, -O-CH-CBH-
OH),

RMN 1% (CDC13), 6 (ppm):
Analyse élémentaire:
3.50 (1 H, dd, J = 3.1 HZ, 1 1.9 Hz, -0-CH- CHH-OH), 2.81 (1 Hl s, 'CH ),
2.41 (2H, m, =CH-CH2-CH-O-), 2.08 (3H, s, OAc), 2.05 (2H, m. -CH(OAc)-CHY) et 1.98 (1 HI S. OH).
calculée: H 7.20 C 64.26; trouvée: H 7.00 C 64.08.
1.14 (2R,3R,SR)-3-acétoxy-5-formy1-2-(Z(E)-pentèn-4-ynyl)- tétrahydrofurane
À une solution de chlorure d'oxalyle (16 PL, 0.18 mmol) dans du CH2CI2
(4 mL), à -78 OC, on ajoute le DMSO (15 PL, 0.21 mmol). On laisse agiter pour
30 minutes puis on ajoute lentement la solution du composé 63 (20.0 mg, 0.09
mmol) dans du CH2C12 (4 mL). On laisse agiter encore une heure puis la Et3N (200 PL) est ajoutée. Le mélange est maintenu à -78 OC pour encore 20
minutes puis on laisse revenir lentement à température ambiante. Le mélange est filtré sur gel de silice puis le solvant est évaporé. Le résidu est purifié par chromatographie éclair en utilisant un mélange Ac0Et:éther de pétrole 35-60 O C (13) pour conduire à l'aldéhyde 64 (19.0 mg, 96%).
C12H1404 Huile incolore
222.24 g 1 mol

RMN 'H (CDC13), 6 (ppm):
RMNI3C (CDC13), 6 (ppm):
Masse exacte m/e:
(ICI isobutane)
0.51 (Ac0Et:éther de pétrole 35-60 OC 1:l)
3270 ( 'CH ), 2920, 2860 (CH aliphati-
ques), 1735 (C=O, aldéhyde), 1430 (CHz), 1245 et 1050 (C-û).
9.70 (1 HI s, CHO),
6.22 (1 Hl td, J = 7.4, 15.9 HZ. GH=Çti-
CH2-1% 5.56 (1 H,d, J = 15.9 HZ, -çII=CH-CH2-), 5.25 (1 HI m, -çtl-OAc), 4.32 (IH, d, J = 10.5 HZ, -O-m-CHO), 4.04 ( l H l ml -O-ÇtL-CH2-CH=),
2.82 (1 HI s, 'CH ),
2.50 (3H, m, -CH(OAc)-ÇtlH- et -O-CH- CH?-CH=) , 2.24 (1 HI d, J = 11 -9 HZ, -CH(OAc)-ÇtLH-), 2.01 (3H, s, OAc).
pour Cl 2H1404 (M+), calculée: 222.0892; trouvée: 222.0900.
SM (IE, 70 eV) 1 33(92), 1 O5 (1 00), 84 (94).

À la solution du complexe borane-sulfure de diméthyle (8.07 mL d'une solution 10 M dans du sulfure de diméthyle, 80.7 mmol) dans du THF (40 mL), a -10 O C (CH30H et glace), on ajoute goutte à goutte le 2-méthyl-2-butène (80.7
mL d'une solution 2 M dans du THF, 0.161 mmol). On laisse le mélange revenir
à température ambiante sur une période de 2 heures puis on refroidit à O OC (bain de glace) et le 1-pentyne (5.0 g, 73.5 mmol) est ajouté goutte à goutte.
Après 30 minutes, on enlève le bain de glace et on laisse le mélange reposer
pour 30 minutes.
Du n-BuLi (32.3 mL d'une solution 2.5 M dans de I'hexane, 80.7 mmol) est refroidi a O O C et la 2,2,6,6-tétraméthylpipéridine (13.6 mL, 80.7 mmol) est
ajoutée goutte à goutte. On laisse le mélange revenir à température ambiante
puis on évapore le solvant. Le résidu est refroidi à O O C et la solution de
disiamyl-1 -pentenylborane, précédemment préparée, est ajoutée via canola. donnant ainsi lieu à une solution orangée lorsque réchauffée à température
ambiante. On refroidit à nouveau à O OC et on ajoute le MesSiCl (15.4 mL, 0.122 mmol). On agite 30 minutes à cette température.
Du AcOH (20 mL) est ensuite ajouté et on chauffe le mélange à 60 OC pour
2 heures. Une fois le résidu refroidi à température ambiante, on ajoute un
mélange de Hz02 30% (44 mL) dans une solution de NaOAc 3 M (200 mL). La
réaction est légèrement exothermique. On laisse reposer 30 minutes puis on
extrait 3 fois avec du pentane. Les extraits organiques combinés sont lavés
successivement avec des solutions de K2CO3 1 M et NaCl saturée, puis évaporés. Le résidu est ensuite dissout dans du pentane puis filtré tout d'abord, sur gel de silice, puis sur alumine. Après évaporation du solvant, le
résidu est purifié par chromatographie éclair en utilisant l'éther de pétrole 35-60
O C comme éluant. Le composé 65 est obtenu dans un rendement de 25%.
C8Hl8Si Liquide incolore

IR (film), v,, (cm1 ):
RMN rH (CDC13). 6 (ppm):
3060 (=C-H), 2950, 2920 (CH aliphati-
ques), 1620 (C=C), 1420 (CHz), 1245 (Si- CH3) et 11 00.
5.63 (1 H, m, -m=CH2),
4.87 (2H. m. -CH=CH2), 1.56 (1 H, m, m-SiMe3), 1.324 -40 (2H, m, CH2), 0.92 (3H, t, J = 7.2 Hz, CH3) et 0.00 (9H, s, TMS).
Les données spectroscopiques de ce composé correspondent à celles
retrouvées dans la littérature?
À la solution de l'aldéhyde 64 (7.1 mg, 0.031 mmol) dans du CH2C12 (2 mL), à -78 OC, on ajoute le complexe borane-trifluoroétherate (10 p l , 0.081
mmol). On ajoute ensuite lentement la solution de Itallylsilane 65 (25.0 mg, 0.176 mmol) dans du CH2CI2 (2 ml). On laisse agiter environ une heure puis on laisse revenir à température ambiante. On ajoute une solution de NaCl saturée (15 mL) puis on extrait avec AcOEt (3x10 mL). La phase organique est
séchée sur Na2S04, filtrée puis évaporée. Le résidu est purifie par chromatographie éclair en utilisant un mélange Ac0Et:éther de pétrole 35-60 O C (3:7) pour conduire a l'alcool 66 (4.9 mg, 52%).

Huile incolore
Rfi
[a]24D:
IR (fi lm), vmax (cm-1 ):
0.39 (Ac0Et:éther de pétrole 3580 O C
3: 7)
3400 (OH), 3280 ('CH ), 2920,2860 (CH
aliphatiques) 1730 (C=O, ester), 1435
(CHz), 1370 (CH3), 1240, IO70 et 1020 (C-
O)-
RMN l H (CDQ), 6 (ppm): 6.19 (IH, td, J = 7.3, 16.0 Hz, =C-CH=CH-), 5.54 (2H, m, CH(0H)-CH2-Ci+=çtL-), 5.34 (1H, m. -==CH-CH2-), 5.21 (IH, m, -m-OAc), 3.85-3.71 (3H, m, -OoM-CH2-CH= et -0-
ul-ai(OH)-CH2), 2.79 (1 H, dl J = 2.1 Hz, 'CH ),
2.43 (2H, rn, -O-CH-CH7-CH=), 2.27 (IH, rn, -CH(OH)-CHH-CH=) 2.21 (2H, m, -CHAH3)
2.1 3-1 -99 (4H, m, -CH(OAc)-CH2- et
-CH(Ok&CH&CH=), 2.03 (3H, s, OAc) et
0.94 (3H, t, J = 7.4 Hz, CH3).
RMN l3C (CDC13), 6 (ppm): 170.3, 141.5, 135.7, 123.9, 111.0, 81.9,
80.0, 79.9, 76.4, 74.3, 71.0, 36.2, 32.6 (e), 25.5, 20.9 et 13.6.
Les données spectroscopiques correspondent à celles retrouvées dans la litterature.e4
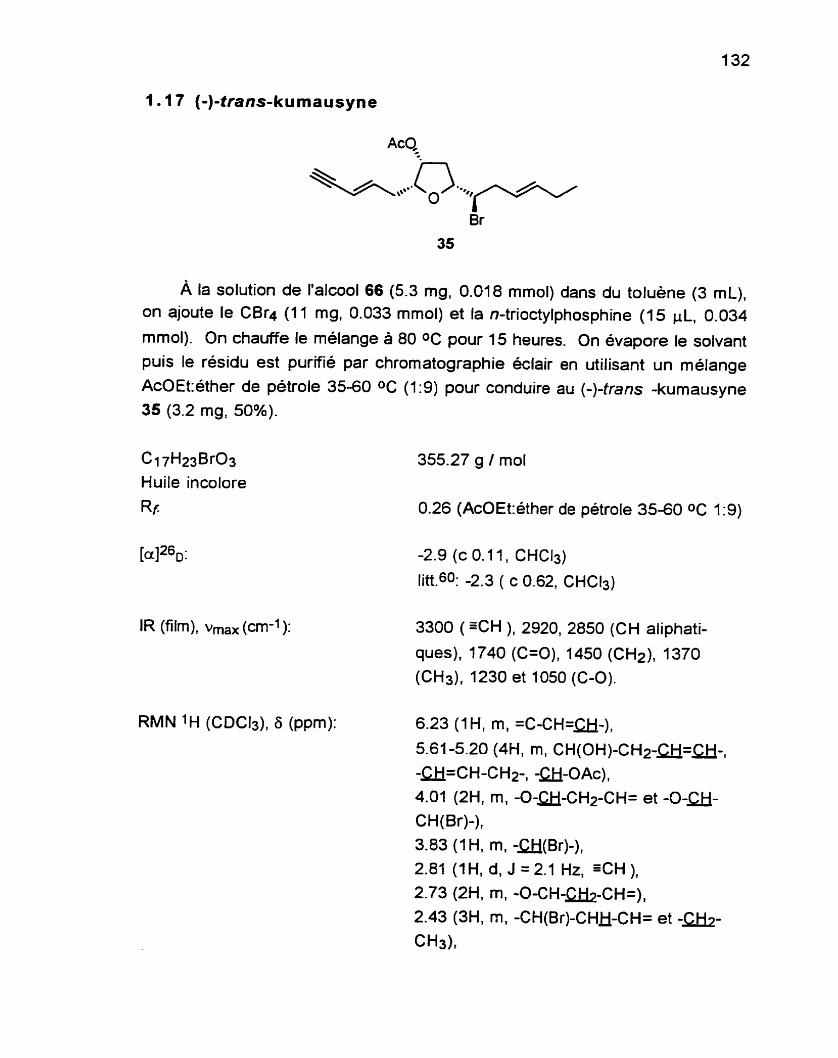
À la solution de l'alcool 66 (5.3 mg, 0.018 mmol) dans du toluène (3 mL), on ajoute le CBr4 (1 1 mg, 0.033 mmol) et la n-trioctylphosphine (1 5 PL, 0.034
mmol). On chauffe le mélange à 80 O C pour 15 heures. On évapore le solvant puis le résidu est purifié par chromatographie éclair en utilisant un mélange
AcOEtéther de pétrole 35-60 OC (1:9) pour conduire au (-)&ans -kumausyne
35 (3.2 mg, 50%).
IR (film), v,, (cm-' ):
355.27 g 1 mol
0.26 (Ac0Et:éther de pétrole 35-60 OC 1:9)
3300 (zCH ), 2920, 2850 (CH aliphati-
ques), 1740 (C=O), 1450 (CHz), 1370 (CH$, 1230 et 1050 (C-O).
RMN 'H (CDCIs), 6 (ppm): 6.23 (1 H, m, =C-CH=çtL-), 5-61 -5.20 (4H, m, CH(0H)-CH2-çtl=m-,
-çtL=CH-CH2-, -çtl-OAC), 4.01 (2H, m, -O-ÇtL-CH2-CH= et -O-=-
CH(Br)-), 3.83 (1 H, m, -çtl(Br)-), 2.81 (1 H, dl J = 2.1 Hz, =CH ), 2.73 (2H. m, -0-CH-CH,-CH=),
2.43 (3H. m, -CH(Br)-CHH-CH= et -CH7-
CH3)l

RMN 1% (CDC13), 6 (ppm):
2.09-2.01 ( 2 H l rn, (0Ac)CH-CH?-) 2.05 (3H. S. OAc), 1.95 (1H. m, -CH(Br)-CHH-CH=) et 0.94 (3H, t, J = 7.5 Hz, CHs).
Les données sont en accord avec celles retrouvées dans la I i t t é r a t ~ r e . ~ ~ ~ ~ ~
Le complexe borane-tétrahydrofurane (1.0 M dans THF, 2.5 mL, 2.50
mmol) est ajouté à la solution du composé 49 (420 mg, 1.13 mmol) dans du THF (35 mL), à O O C , sur une période de 30 minutes. On laisse alors agiter à
température ambiante pour toute la nuit. On ajoute ensuite un peu d'eau, jusqu'à ce qu'il cesse d'y avoir dégagement de Hz. On effectue plusieurs CO-
évaporations avec une solution de 2% d'AcOH dans du MeOH, puis avec du benzène afin d'éliminer l'eau. Le résidu est purifié par chromatographie éclair

en utilisant comme éluant un mélange AcOEt:CH2C12 (2:8) pour conduire au lactol 79 (273 mg, 65%), sous forme d'un mélange d'isomères.
C21H2804Si Huile incolore
Rt.
372.54 g f mol
0.20 (AcOEt:CH2C12 2:8)
IR (film), vmax (cm-1):
RMN 1H (CDC$), 6 (ppm):
RMN13C (CDCI3), 6 (pprn):
Analyse élémentaire:
3500-3220 (OH), 3060 (=CH aromatiques),
2920, 2850 (-CH aliphatiques), 1465, 1420
(CHz), 1 4 05 et 1 035 (C-O).
7.71 (4H, m, H aromatiques),
7.43 (6H. rn, H aromatiques),
5.42 [S. 181 (1 H, m [dd (J = 9.1 Hz)], (HO)-
m-O-), 4.49 [4.37] (1 H, m, CH2-=-OH),
4.25 [4.00] (1 H, m, -O-U-CH2).
3.84-3.48 (2H,m, CH-CHPOTBDPS),
2.70 (2H, m, OH), 2-31 -2.14 [2.02-1.861 (2H, ml (0H)CH-) et
1.10 [ A .O81 (9H, S. t-butyle).
calculée: H 7.58 C 67.71 Si 7.54; trouvée: H 7.73 C 67.64 Si 7.69.

2.2 (1 R, 3s)-1,3-dihydroxy-1 -(t-butyIdiph6nylsilyloxym6thyl)-4- pentène
À la suspension de t-BuOK (55 mg, 0.46 mmol) dans de l'éther (25 mL), on
ajoute le CH3PPh3Br sec (171 mg, 0.48 mmol), a température ambiante. On laisse agiter 2 heures puis on ajoute le lactol 79 (90 mg, 0.24 mmol) en solution
dans de l'éther (5 mL). Le mélange est agité toute la nuit à température ambiante. On ajoute alors un peu d'eau et on extrait plusieurs fois avec un mélange Et2O:éther de pétrole 35-60 O C (21). Les phases organiques réunies sont séchées sur Na2S04, filtrées puis concentrées. Le résidu est purifié par chromatographie éclair en utilisant comme éluant un mélange Et2O:éther de pétrole 35-60 O C (1 : 1) pour conduire au di01 80 (58 mg, 65%).
C22H3003Si Huile incolore
Rt:
RMN 1H (CDCI3), 6 (ppm):
370.56 g / mol
0.24 (Et2O:éther de pétrole 35-60 OC 1 : 1 )
3490-3240 (OH), 3070, 3040 (CH aroma-
tiques), 2930, 2850 (CH aliphatiques), 1465, 1420 (CHz), 1 1 O5 et 1070 (C-O).
7.65 (4H, m, H aromatiques),
7.42 (6H, m, H aromatiques), 5.86 (11-1, ddd, J = 5.9, 10.5, 17.1 Hz, H2C=çtl-CH(OH)), 5.26 (1 H l d, J = 17.3 Hz, HBC=CH-), 5.09 (1 HI d, J = 10.5 HZ, HHC=CH-),
4.37 (1 H, rn, H2C=CH-çtL(OH)),

RMN 13C (CDC13), 6 (ppm):
Analyse élémentaire:
3.99 (1 HI ml (OH)=-CH2-OTBDPS), 3.63 (1 HI dd, J = 4.0, 10.1 HZ, CH-CHH- OTBDPS). 3.53 (1 H, dd. J = 7.1 , 10.1 HZ, CH-CHH- OTBDPS), 3.18 (1 HI SI, OH), 2.96 (1 H, SI, OH), 1.60 (2H. m, (0H)CH-CH7-CH(0H)) et 1 -06 (9H, s, t-butyle).
calculée: )-1 8.1 6 C 71 -31 ; trouvée: H 8.22 C il. 1 1.
2.3 (1 R, 3S)-1,3-dihydroxy-l-(t-butyldiphénylsilyIoxyméthyl)-4- pentène
A la solution du composé 57 (27 mg, 0.73 mmol) dans un mélange de THF (2 mL) et de MeOH (0.2 mL), à -78 OC, on ajoute le EtzBOMe (12 PL, 0.87
mmol). On laisse agiter 15 minutes puis on ajoute le NaBH4 (3 mg, 0.079 rnmol). On laisse le mélange agiter 4 heures à cette température puis on ajoute
du AcOH (0.2 mL). On ajoute alors un peu d'une solution aqueuse saturée de NaHCO3 (1 0 mL) et on extrait plusieurs fois avec de I'AcOEt (3x1 0 mL). Les
phases organiques réunies sont ensuite lavées avec une solution aqueuse saturée de NaCI, séchées sur Na2S04, filtrées puis concentrées. On ajoute alors un peu de MeOH (10 mL), on agite puis on évapore à nouveau le solvant. Cette opération est répétée cinq fois. Le résidu est purifié par chromatographie

éclair en utilisant comme eluant un mélange Ac0Et:ether de pétrole 35-60 OC (2:8) pour conduire au diol 80 (25 mg, 92%) dont les caractéristiques ont été
énumérées auparavant.
.'#,/
OTBDPS
On place dans un tricol le PdCI2 (8.9 mg, 0.05 mmol), le CuC12 (261 mg,
1.53 mrnol) et le NaOAc (167 mg, 2.04 mmol). On y ajoute ensuite la solution
du diol 80 (190 mg, 0.51 mmol) dans de I'AcOH (50 mL). On fait barbotter du CO dans la solution, qui passe initialement d'une couleur verte à une couleur
rouille. On laisse le mélange agiter ainsi pour 24 heures. Suite à cette période,
le mélange est dilué dans du benzène puis filtre sur cellulose. La phase organique recueillie est lavée successivement avec des solutions de NaHC03
saturée et NaCl saturée. Une huile est obtenue après séchage de la phase organique sur MgS04, filtration et évaporation. Ce résidu est ensuite
chrornatographié selon la technique éclair en utilisant comme éluant un mélange Et20:éther de pétrole 35-60 O C (6:4) pour conduire au bicycle 81 (1 82
mg, 90%).
C23H2804Si Huile incolore
R f i
IR (film), vmax (cm-'):
396.56 g / mol
0.29 (Et2O:éther de pétrole 35-60 OC 6:4)
3060, 3040 (CH aromatiques), 2920, 2850
(CH aliphatiques), 1780 (C=0, lactone),

RMN l H (CDCb), 6 (ppm):
RMN 1% (CDCIJ), 6 (ppm):
Analyse élémentaire:
1460. 1420 (CH2). 1 175,1140.1 105 et 1070 (C-O).
7.66 (4H, ml H aromatiques),
7.40 (6H. ml H aromatiques), 5.12 (1 H, m, -CO-O-çli-), 4.82 (1 HI ml -CO-CH2-ÇtL-), 4.30 (1 H, ml ÇtL-CH2-OTBDPS), 3.81 (1 HI dd, J = 3.6, 11.1 HZ, CH-CHH- OTBDPS), 3.64 (1 Hl dd, J = 3.6, 1 1.1 HZ, CH-ÇtlH- OTBDPS), 2.70 (2H, m, -CO-m-CH-) 2.35 (1 Hl m, -CHH-CH-CH2-OTBDPS), 2.19 (IH, ml -mH-CH-CH2-OTBDPS) et 1 .O5 (9H, s, t-butyle).
calculée: H 7.12 C 69.66;
trouvée: H 7.1 8 C 69.78.
2.5 (lS,3RS,SS,7R)-2,6-dioxa-7-(t-butyldiphényfsilyloxymethyl)- bicyclo [3.3.0] octan-3-01
A une solution du bicycle 81 (100 mg, 0.25 mmol) dans du THF (5 mL) à -78 OC, on ajoute lentement le DIBAL-H (1.0 M dans du toluène, 380 PL, 0.38

mmol). On laisse agiter a cette température pour 3 heures puis on ajoute un peu d'une solution de NH4CI saturée. On laisse le mélange revenir à
température ambiante. Par la suite, on ajoute un peu d'une solution de HCI 10% et de l'éther, et la solution est agitée fortement pendant quelques minutes. On effectue plusieurs extractions à l'éther et les phases organiques réunies
sont lavées avec une solution saturée de NaHC03 puis avec une solution de
NaCl saturée. Après séchage des phases organiques sur Na2S04, filtration et concentration, le résidu est purifié par chromatographie éclair en utilisant comme éluant un mélange AcOEt:CH2C12 (595) pour conduire au lactol 82 (90
mg, go%), sous forme d'un mélange d'isomères.
C23H3004Si Huile incolore
398.57 g I mol
0.30 (AcOEt:CH2C12 595)
IR (film), vmax (cm-'):
RMN 'H (CDC$), 6 (ppm):
3400 (OH), 3050, (CH aromatiques). 2920,
2850 (CH aliphatiques), 1465, 1420 (CH2)
et 11 05 (C-O).
7.66 (4H, m, H aromatiques),
7.39 (6H, ml H aromatiques), 5.65 [5.44] (1 Hl m, -çtl(OH)-), 4.83 (1 Hl m, -CH(OH)-CH2-ÇtL-),
4.72 [4.66] (1 H, ml -CH(OH)-O-ÇtL-), 4.50 [4.16] (1 Hl ml -O-ÇtL-CH2-OTBDPS),
3.73 [3.61] (2H, m, -O-CH-CHPOTBDPS), 3.03 (1 Hl SI, OH),
2.35-1 -85 (4H, m. (0H)CH-CH?-CH- et
-C H7-CH-C Hz-OTBDPS) et 1.05 (9H, s, t-butyle).
RMN '3C (CDCIJ), 6 (ppm): 135.5 [135.5], 133.4 [133.3], 129.6 [129.5],
1 27.6 [l27.5], 99.9 [99.7], 85.6 [83.2], 82.9

Analyse élémentaire: calculée: H 7.59 C 69.31; trouvée: H 7.79 C 68.97.
A la suspension de t-BuOK (285 mg, 2.50 mmol) dans de l'éther (65 mL),
on ajoute le bromure de propyltriphénylphosphonium sec (970 mg, 2.52 mmol),
à température ambiante. On laisse agiter 2 heures puis on ajoute le lactol 82
(200 mg, 0.50 mmol) en solution dans de l'éther (20 mL). Le mélange est agité toute la nuit à température ambiante. On ajoute alors un peu d'eau et on extrait
plusieurs fois avec un mélange Et2O:éther de pétrole 35-60 OC (23). Les phases organiques réunies sont séchées sur N a ~ S 0 4 ~ filtrées puis
concentrées. Le résidu est purifié par chromatographie éclair en utilisant comme éluant un mélange Et2O:éther de pétrole 35-60 OC (25:75) pour conduire au composé 83 (187 mg, 88%).
C26H3603Si Huile incolore
R f:
424.65 g I mol
0.35 (Et2O:éther de pétrole 35-60 O C
25:75)

IR (film), v,, (cm-'):
RMN 1H (CDC13), 6 (ppm):
3360 (OH), 3040, 3020 (CH aromatiques),
2940, 291 0, 2830 (CH aliphatiques), 1460,
1440, 147 5 (CH2), 1 100 (C-û).
7.68 (4H, ml H aromatiques),
7.39 (6H, m, H aromatiques),
5.48 (1 H, ml JI = 7.0 Hz, CH3-CH2-CH=), 5.39 (1 H, m. J1 = 7.0 Hz, CH3-CH2-
C H =a-), 4.34 (1 H, m. -O-ÇtL-CH2-OTBDPS), 4.27 (1 Hl ml (OH)-=-CH2-), 3.87 (lH, td, J = 2.7, 7.3 HZ, -CH-CH(0H)-), 3.74 (IH, dd, J ~ 4 . 2 , 10.8 HZ, -CH-ÇHH- OTBDPS), 3.63 (1H, dd, J ~ 4 . 2 , 10.8 HZ, -CH-ÇHH- OTBDPS), 2.41 (2H, ml =CH-CHzCH-O-),
2.09 (4H, m. CH3-CHPCH= et (HO)- CH-
U-), 1.57 (1 H, SI, OH), 1 -04 (9H, s, t-butyle) et
0.96 (3H, t, J = 7.5 HZ, -CH3).
RMN 1% (CDCI3), 6 (ppm): 135.6, 134.1, 133.6, 129.6, 127.6, 124.3,
82.5, 73.3, 66.3, 37.2, 32.5, 27.2, 26.9,
20.7, 19.2 et 14.2.
Analyse élémentaire: calculée: H 8.54 C 73.54; trouvée: ti 8.25 C 73.68.

2.7 (2s. 3S, SR)-2-hydroxy-3-pentyl-5-(t-butyldi phénylsilyloxy- méthyl)-tétrahydrofurane
Le composé 83 (162 mg, 0.38 mmol) dans de I'hexane (40 mL) est placé en présence de Pd I C (50 mg) sous 45 psi de Ha. Le mélange est ainsi agité
fortement pour 15 heures. Le mélange est filtré sur célite puis le solvant est
évaporé. Le résidu est purifié par chromatographie éclair en utilisant comme éluant un mélange Ac0Et:éther de pétrole 3540 O C (25:75) pour conduire au
composé 84 (1 56 mg, 96%).
C26H3803si Huile incolore
R 6
426.67 g l mol
0.35 (Ac0Et:éther de pétrole 3540 O C 25:75)
3360 (OH), 3040, 3020 (CH aromatiques),
2900, 2830 (CH aliphatiques), 1450, 141 5 (CHz), 1 100 (C-0).
RMN AH (CDCh), 6 (ppm): 7.72 (4H, ml
7.42 (6H, rn, 4.35 (1 H, ml 4.25 (1 H, ml 3.86 (IH, rn,
H aromatiques),
H aromatiques),
-O-CH-CH?-OTBDPS),
(OH)-la-CH2-), -çtL-CH(0H)-),
3.68 (AH, dd, J = 3.9, 10.8 HZ, -CH-CHH- OTBDPS), 3.54 (1 H, dd, J = 3.9, 10.8 HZ, -CH-CHH-
OTBDPS), 2.09 (1 H, m, -O-CH-çtLH-CH2-),

1.93 (IH, m. -O-CH-çtLH-CH2-), 1.59-1.25 (8H, rn, CH3-(CHp)3- et (OH)- CH-cfi7-) 1.18 (1 H, SI, OH), 1 -04 (9H, S. t-butyle) et 0.97 (3H, t, J = 7.4 HZ, -CH3).
RMN 13C (CDC13). 6 (ppm): 135.5, 133.5, 129.5, 127.5, 82.8, 76.9,
73.4, 66.2, 37.1, 31.9, 28.9, 26.7, 26.1, 22.5, 19.1 et 13.9.
Analyse élémentaire: calculée: H 8.98 C 73.19; trouvée: H 9.16 C 73.01.
2.8 (2S, 3S, SR)-3-acétoxy-Z-penty1-5-(t-b~ tyldiphénylsilyloxy- méthyI)-tétrahydrofurane
À la solution du composé 84 (18.0 mg, 0.042 mmol) dans de la pyridine (2 mL), on ajoute le DMAP (1 .O mg, 0.008 mmol) et l'anhydride acétique (25~1,
0.27 mmol). On laisse agiter pour deux heures a température ambiante. Si la
réaction ne semble pas complète après ce temps, on peut ajouter quelques gouttes d'anhydride acétique. Le mélange est ensuite dilué avec de l'éther et
la phase organique est lavée successivement avec des solutions de HCI IO%, NaHC03 saturée et NaCl saturée. La phase organique est séchée sur MgS04, filtrée puis évaporée. Le résidu est purifie par chromatographie éclair en utilisant un mélange Et2O:éther de pétrole 35-60 O C (15:85) pour conduire à l'acétate 85 (19.7 mg, 100%).
C28H4004Si 468.71 g 1 mol

Huile incolore
Rt:
[a]24D:
IR (film), v,, (cm-'):
RMN H (CDC$), 6 (pprn):
RMN I3C (CDC13), 6 (ppm):
Analyse élémentaire:
0.37 (Et2O:éther de pétrole 35-60 OC
1 5:85)
3040, 3020 (CH aromatiques), 2940, 2900,
2830 (CH aliphatiques), 1725 (C=O, ester), 1450, 141 5 (CHz), 1355, 1225, 1100 (C-
o 1.
7.71 (4H, m. H aromatiques),
7.66 (6H, m, H aromatiques),
5.35 (IH, m, -çLI(OAc)), 4.31 (1 Hl m. -O-çtl-CH2-OTBDPS), 4.00 (1 Hl m, (0H)m-CH2-CH2),
3.80 (IH, dd, J = 3.9, 10.8 HZ, -CH-ÇtLH- OTBDPS), 3.64 (1 H, dd, J = 3.9, 10.8 HZ, -CH-WH-
OTBDPS), 2.33 (1 H, m, -CH(OAc)-ÇtLH-), 2.1 1 (3H, s, OAc), 2.05 (1 H, rn, -CH(OAc)-mH-), 1.59-1.49 (4H, m, -O-CH-CH7-Cb-), 1.30 (4H, rn, CH3-CH7-CH2-), 1 .O6 (9H, s, t-butyle) et 0.96 (3H, t, J = 7.5 HZ, -CH3).
calculée: H 8.60 C 71 -75; trouvée: H 8.75 C 71.78.

2.9 (2S, 3s. 5R)-3-ac6toxy-2-pentyl-5-hydroxyméthyl-t~trahydro- furane
A la solution de l'acétate 85 (93 mg, 0.20 mmol) dans du THF (15 mL). on ajoute le TBAF (1 -0 M dans du THF, 300 PL, 0.30 mmol). On laisse agiter pour
deux heures à température ambiante. Si la réaction ne semble pas complète après ce temps, on peut ajouter quelques gouttes de TBAF. On évapore le solvant puis le résidu est purifié par chromatographie éclair en utilisant un
mélange Ac0Et:éther de pétrole 35-60 OC (1:l) pour conduire a l'alcool 86
(40.5 mg, 88%).
230.30 g I mol
0.24 (Et2O:éther de pétrole 35-60 OC 1 : 1 )
IR (film), vmax (mm'): 3520-3300 (OH), 2950,2920, 2860 (CH
aliphatiques), 1735 (C=Ol ester), 1450 (CHz), 1370, 1240, 1030 (C-O).
RMN 1H (CDCh), 6 (ppm): 5.33 (1 Hl m, -Çtl(OAc)), 4.29 (1 H, m, -O-m-CH2-OTBDPS), 3.91 (1 H, mi (OH)-m-CH2-CH2).
3.71 (1 H, m, -CH-mH-OTBDPS), 3.50 (1 H, ml -CH-mH-OTBDPS),
2.06 (3H, s, OAc), 2.00 (1 H, m, -CH(OAc)-UH-), 1.85 (1 H, SI, OH), 1.57-1 2 4 (9Hi ml 4xCH2- et -CH(OAc)- çt[H-) et

0.87 (3H, t, J = 7.5 Hz, CH3).
RMN 1% (CDC13), 6 (ppm):
Masse exacte mie: (IC, isobutane)
SM (IE. 70 Ev)
pour C12H2304 (MH+), calculée: 231.1596;
trouvée: 23 1 .1 600.
La suspension de KH (22.4% dans l'huile, 3.17 g, 17.80 mmol) est placée dans un ballon, sous N2. On lave le KH avec deux portions d'hexanes puis on laisse passer un courant d'azote pour éliminer les dernières traces de solvant. Une fois le KH lavé et séché, on ajoute rapidement le 1,3diaminopropane
fraîchement distillé (20 mL) et on agite vigoureusement. On refroidit si
nécessaire dans un bain de glace pour éviter qu'il n'y ait formation excessive
de mousse. On note une coloration bleue au début de la réaction puis elle disparaît pour laisser place à une coloration jaune-brun. On laisse ensuite agiter 2 heures à la température ambiante afin d'obtenir le KAPA. Au 3-nonyn- ol (250 mg, 1.78 mmol), placé dans un ballon sous N2, on ajoute, via canula, le KAPA préparé préalablement. On agite vigoureusement pendant 15 heures à
température ambiante. Le mélange est ensuite jeté dans de l'eau glacée et extrait avec de I'hexanes (4 fois). Les phases organiques réunies sont lavées
sucessivernent avec des solutions de HCI IO%, NaHC03 saturée et NaCl saturée. Après séchage sur MgS04. filtration et évaporation, le résidu est purifié par distillation sous vide. On recueille 200 mg (80%) du composé 88.
CgH160 140.22 g 1 mol

Liquide incolore P.é:
IR (film), vm, (cm-'):
136-1 38OC (30 mm de Hg)
1itt.l g: 83 OC (0.05 mm de Hg)
0.19 (AcOEt: CH2Ci2 1 S:85)
3280 (OH), 291 0, 2840 (CH aliphatiques),
2090 (CEC ), 1450, 1410 (CHz), 1040 (C-
O).
3.63 (2H, t, J = 6.6 HZ, XHPOH),
2.16 (2H, td, J = 2.6, 6.6 HZ, =C-ÇtL-), 1.91 (IH, t , J = 2.6 HZ, HC=C-), 1.55 (4H. ml -(CH&OH) et
1.28 (6H. m, -(CH2)3).
Les données pour ce composé sont en accord avec celles retrouvées dans la littérature1 l 9 .
On place, sous atmosphère de H2, l'alcyne 88 (100 mg, 0.71 rnmol), la quinoline (1 goutte) et le catalyseur de Lindlar (Pd i CaC03) (20 mg) dans 15
mL d'un mélange hexanes:l-pentene 6:1. On laisse agiter 30 minutes à
température ambiante puis on filtre le mélange sur célite. On lave ensuite successivement avec une solution d'acide acétique 1 O%, NaHC03 saturée puis
NaCl saturée. Après séchage sur NaZS04, filtration et évaporation, le résidu est purifié par chromatographie en utilisant un mélange AcOEt:CH2CI2 (1 5:85) pour conduire à l'alcène 89 (91 mg, 90%).

CgHiaO Liquide incolore P-é:
Rf:
IR (film), v,, (cm-1):
RMN~H (CDC13), 6 (ppm):
RMNI~C (CDC13), 6 (ppm):
142.24 g I mol
128-130 OC (22 mm de Hg) litt.121: 1 13-1 23 O C (2 mm de Hg)
3280 (OH), 2900, 2830 (CH aliphatiques),
1630 (C=C), 1450 (CH2). 1 040 ( C a ) .
5.79 (1 H, ddt, J = 17.1, 10.2, 6.7 Hz, CH2=
ai-), 4.95 (2H, ml CH7=CH-), 3.61 (2H, t, J = 6.7 HZ, -C&-OH), 2.02 (2H, m. CH~=CH-CHP-), 1.52 (3H1 ml -m-CH2-OH et OH), 1.31 (8H, m, -(CH2)4-).
A la solution du composé 89 (430 mg, 3.02 mmol) et du CBr4 (1.107 g, 3.34 rnmol) dans du CH2CI2 (1 0 ml), on ajoute la ?Ph3 (895 mg, 3.32 rnmol) par petites portions. On laisse agiter pendant deux heures à température ambiante. Le solvant est évaporé et le résidu est placé dans un mélange Et2O:éther de pétrole 35-60 O C (1 : 1 ) afin de faire précipiter les sous-produits de la réaction, qu'on élimine par filtration. Après évaporation du solvant, le résidu est alors purifié par chromatographie éclair en utilisant de I'hexanes pour conduire au bromure 90 (465 mg; 75%).

C9H17Br Liquide incolore
R fi
IR (film), vmax ( c d ):
RMN'H (CDC13), 6 (ppm):
205.1 4 g / mol
0.60 (Hexanes)
3050 (CH alcène), 2900, 2930 (CH alipha-
tiques), 1625 (C=C), 1445 et 1420 (CH2).
5.79 ( IH, ddt, J = 17.1, 10.2, 6.7 Hz, CH2=
ai-), 4.96 (2H. m, CH?=CH-) 3.39 (2H, t, J = 6.8 HZ, -(&-Br)* 2.01 (2H, m, CH~=CH-CHP),
1.84 (2H, q, XH7-CHz-Br), 1.36 (8H, m, -(CH2)4-).
RMN13C (CDC13), 6 (ppm): 138.9, 114.1, 33.8, 33.6. 32.7, 28.8, 28.7,
28.5 et 28.0.
Les données spectroscopiques de ce composé sont en accord avec celles retrouvées dans la littérature. l 23

3.1 Alkylation de la butyrolactone 102
À une solution de LDA (1.5 M dans du cyclohexane, 2.2 mL, 3.30 mmol) et de HMPA (600 PL, 3.45 mmol) dans 50 mL de THF, à -50 OC, on ajoute
lentement la solution du composé 102 (1.0 g, 2.80 mmol) dissout dans 20 mL de THF. On laisse agiter une heure à cette température puis on ajoute l'iodure d'éthyle (300 pL, 3.75 mmol). On laisse agiter encore 15 heures à -50 OC puis
on ajoute un peu d'une solution de NH4CI saturée et de l'éther. On laisse le
mélange revenir à température ambiante et on procède ensuite à plusieurs
extractions avec de l'éther. Les phases organiques réunies sont lavées successivement avec HCI 1 O%, NaHS03 saturée, NaHC03 saturéee et
finalement NaCl saturée. Après séchage des phases organiques sur MgS04, filtration et concentration, on obtient un mélange de deux diastéréoisomères
(655 mg, 61%) dans un rapport 5.2:1, tel que déterminé par RMN 1H. Les
isomères peuvent être sépares par chromatographie éclair en utilisant comme
éluant un mélange Et2O:hexanes (3:7). Le composé 103 est le produit
majoritaire.
C23H3003Si
Huile incolore
RF
382.57 g 1 mol
0.28 (Etno: hexanes 3:7)
20.1 (C 1 .O4, CHC13)

IR (film), v,, (an-1):
RMN1H (CDC13), 6 (ppm):
RMNf3C (CDC13), 6 (ppm):
3080, 3060 (CH aromatiques), 2970, 2940,
2870 (CH aliphatiques), 1780 (C=0, lac- tone), 1465. 1435 (CH2). 1 1 75 et 1 1 15 (C-
O).
7.65 (4H, m. H aromatiques),
7.41 (6H, m, H aromatiques), 4.53 (1 H, m, CH-CH2-OTBDPS), 3.84(1H, dd, Jz3.4 , 11.1 HZ, CH-CHH- OTBDPS), 3.66 (IH, dd, J = 3.2, Il .i HZ, CH-CHH- OTBDPS), 2.71 (1 HI m. C-CH2-CH3). 2.36 (IH, m, Et-CH-CHH-), 2.00 (IH, m, Et-CH-CHH-), 1.89 (1 HI m, C&LCH3), 1.48 (1 HI m, CHH-CH3), 1 .O4 (9H, s, t-butyle) et
1.02 (3H, t, J = 7.4 Hz, CH3).
C23H3003Si
Huile incolore
Rfi
382.57 g I mol

[a]*eD:
IR (film), vwx(m-~ ) :
RMN'H (CDCi3), 6 (pprn):
R M N W (CD&), 6 (pprn):
3080, 3060 (CH aromatiques). 2980, 2950,
2870 (CH aliphatiques), 1780 (C=O, lac- tone), 1480, 1435 (CHz), 1 180 et 1 120 (C-
O)-
7.66 (4H, m, H aromatiques),
7.40 (6H, ml H aromatiques), 4.45 (1 Hl ml C&CH2-OTBDPS), 3.85 (1 Hl dd, J = 3.6, 11 -2 HZ, CH-CBH- OTBDPS), 3.72 (AH, dd, J ~ 4 . 3 , 11.2 HZ, CH-CYH- OTBDPS), 2.59 (1 HI ml CH-CH2-CHJ), 2.30 (1 H l ml Et-CH-CHH-), 1.91 (2H, m, Et-CH-CHH- et CHH-CH3), 1.49 (1 HI ml CHH-CH3), 1 .O5 (9H, s, t-butyle) et 0.98 (3H, t, J = 7.4 Hz, CH3)-

3.2 Hydroxylation des butyrolactones 103 et 104
A la solution de LiHMDS (1 .O M dans du THF. 5.1 mL, 5.1 0 mmol), à -78
O C , on ajoute lentement le mélange de composés 103-104 (980 mg, 2.56
mmol) dans du THF (20 mL). On laisse agiter une heure -78 O C puis on ajoute le MoOPH (2.22 g. 5.12 mmol). La solution est ensuite agitée pour 3 heures additionnelles a cette température. On ajoute alors un peu d'une solution de Na2SO3 saturée et de I'éther puis on laisse le mélange revenir à
température ambiante. On agite fortement la solution et on ajoute suffisamment d'eau pour dissoudre tous les sels. On évapore la majeure partie du THF puis on extrait ensuite le résidu plusieurs fois à l'éther. Les phases organiques réunies sont lavées avec une solution de HCI 10%. puis avec NaHC03 saturée puis enfin avec une solution de NaCl saturée. Après séchage des phases organiques sur MgS04, filtration et concentration, le résidu est purifié par chromatographie éclair avec comme éluant un mélange Et2O:éther de pétrole 35-60 OC:CHCl3 (45:50:5) pour conduire majoritairement au composé 105
(544 mg, 53%). de même qu'une petite quantité de son épimère en C-2 106
(102 mg, 10%). Le composé 106 est recristallisé dans de I'hexane.
C23H3004Si Huile incolore
R t.
398.57 g / mol
0.23 (Et20: hexane: CHCI3 3MO: 1 5)
IR (film), v,,, (cm-' ): 3520-3300 (OH), 3070 (CH aromatiques),
2970. 2940, 2870 (CH aliphatiques), 1770

RMN1 H (CDC13), 6 (ppm):
RMN13C (CDC13), 6 (ppm):
Analyse élémentaire:
(C=O, lactone), 1465, 1435 (CHz), 1205,
1130 et 1115 (C-O).
7.66 (4H, m, H aromatiques),
7.41 (6H, ml H aromatiques), 4.71 (1 H, rn, CH-CH2-OTBDPS), 3.92(1H, dd, J =3.2, 11.5 HZ, CH-CHH-
OTBDPS), 3.70 (1 H, dd, J = 3.7, 11 -5 HZ, CH-CHH-
OTBDPS), 2.27 (IH, dd, J = 8.6, 13.6 HZ, Et-CH-
CHW, 2.89 (IH, SI, OH),
2.16 (AH, dd, J = 6.7, 13.6 HZ, Et-CH-
WH-), 1.87 (IH, m, CHH-CHd. 1.72 (1 H, m, CHH-CH3), 1.04 (9H, s, t-butyle) et
0.99 (3H, t, J = 7.4 Hz, CH3).
calculée: H 7.59 C 69.31;
trouvée: H 7.78 C 69.23.

C23H3004Si Solide blanc
Rfi
RMNlH (CDCI3), 6 (ppm):
398.57 g I mol
0.29 (Et20: hexane: CH CI3 35:5O: 1 5)
91-92 O C (hexanes)
3530-3300 (OH), 3090, 3060 (CH aromati-
ques), 2950, 2870 (CH aliphatiques), 1775 (C=O, lactone), 1480, 1465, 1435 (CHz), 1205, 1135 et 1115 (C-0).
7.66 (4H, m, H aromatiques),
7.43 (6H, m, H aromatiques), 4.48 (? Hl m, CH-CH2-OTBDPS), 3.90 (1 H, dd, J = 3.0, 1 1.6 HZ, CH-CBH- OTBDPS), 3.68 (1 H, dd, J = 3.7, 11 -6 HZ, CH-CHH- OTBDPS), 3.15 (lHl SI, OH), 2.39 (IH, dd, J = 7.4, 13.4 HZ, Et-CH-
CH+-) , 2.28 (lH, dd, J =7.0, 13.4 HZ, Et-CH-
m H -1 1
1-83 (1 HI m, CHH-CH3), 1.72 (1 H, rn, CHH-CHs),

RMN13C (CDCIJ), 6 (ppm):
1.05 (9H. s, t-butyle) et 1.03 (3H, t, J = 7.4 Hz, CH3).
Le complexe borane-tétrahydrofurane (1 .O M dans THF, 10.0 mL, 10.00
mmol) est ajouté à la solution du composé 105 (785 mg, 1.97 mmol) dans du THF (60 mL), a O OC, sur une période de 30 minutes. On laisse alors agiter a température ambiante pour toute la nuit. On ajoute ensuite un peu d'eau,
jusqu'à ce qu'il cesse d'y avoir dégagement de Hz. On effectue plusieurs CO-
évaporations avec une solution de 2% d'AcOH dans du MeOH, puis avec du
benzène afin d'éliminer l'eau. Le résidu est purifié par chromatographie éclair
en utilisant comme éluant un mélange Et2O:hexanes (6:4) pour conduire au
lactol 1 O7 (677 mg, 86%), sous forme d'un mélange d'isomères.
C23H3204Si Huile incolore
R f i
400.59 g 1 mol
0.1 4 (Et2O:hexane 6:4)

IR (film), vmax (an-1):
RMNIH (CDCI3), 6 (ppm):
3540-3230 (OH), 3080, 3060 (CH aromati-
ques), 2940, 2870 (CH aliphatiques), 1460, 1430 (CH2), 1 1 15 et 1050 (GO).
7.66 (4H, m, H aromatiques),
7.40 (6H, m, H aromatiques), 5.10 [4.93] (1 HI m [d, J = 7.8 Hz], -O-- OH). 4.41 (1 HI m, -m-CH2-OTBDPS), 3.84-3.70 [3.64-3.441 (2H, m, GH7- OTBDPS), 2.26 (2H, SI, OH), 2.09-1 -60 (4H, m, 2x -CH?), 1-07 [1.04] (9H1 s, t-butyle) et
0.98 (3H, t, J = 7.4 Hz, CHj).
RMN13C (CDCI3), 6 (ppm): 135.6 [135.5], 133.3 [132.2], 129.9 (129.51,
127.7 [127.5], 102.4 [100.9], 84.5 [80.2], 78.9 [77.1], 65.5 [65.1], 38.7 [35.5], 30.5 [27.4], 26.7 126.71, 19.1 [19.0] et 8.2 [8.0].
Analyse élémentaire: calculée: H 8.06 C 68.96; trouvée: H 8.14 C 68.97.

À la suspension de t-BuOK (1.1 1 g, 9.24 mmol) dans de l'éther (250 mL), on ajoute le bromure de méthyltriphénylphosphonium sec (3.30 g, 9.24 rnmol),
à température ambiante. On laisse agiter 2 heures puis on ajoute le lactol 107 (373 mg, 0.93 mmol) en solution dans de l'éther (10 mL). Le mélange est agité
toute la nuit à température ambiante. On ajoute alors un peu d'eau et on extrait
plusieurs fois avec un mélange Et2O:éther de pétrole 3540 O C (23). Les phases organiques réunies sont séchées sur Na2S O 4 , filtrées puis
concentrées. Le résidu est purifié par chromatographie éclair en utilisant
comme éluant un mélange Ac0Et:hexanes (15:85) pour conduire au diol 108
(251 mg, 68%).
C24H3403Si
Huile incolore
Rfi
IR (film). vmax(cm-l):
R M N ~ H (CDC13), 6 (ppm):
398.62 g / mol
0.17 (AcOEt: hexanes 15:85)
4.2 (c 1.04, CHC13)
-1 1.1 (C 1.32, EtOH)
3500-3300 (OH), 3090 (CH aromatiques),
2950, 2870 (CH aliphatiques), 1475, 1465,
1435 (CH2), 1 120 et 1070 (C-O).
7.65 (4H, rn, H aromatiques),
7.41 (6H, ml H aromatiques), 5.81 (IH, dd, J = 10.8, 17.4 HZ. CH2=çtL-),
5.21 (1 Hl d, J = 17.4 Hz, CHH=CH-), 5.08 (1 H, d, J = 10.8 HZ, CHH=CH-),

RMNl3C (CDC13), 6 (ppm):
Analyse élémentaire:
4.06 (1 H, m, a(OH)-CH2-OTBDPS), 3.57 (W, m. -CH34TBDPS), 3.00 (1 H, SI, OH), 2.95 (1 H, SI, OH), 1.61 (4H, m. -CH?-CH3 et CHAH(0f-i)-
CH2-OTBDPS), 1.06 (9H, s, t-butyle) et
0.84 (3H. t. J = 7.5 HZ. -CH2-CH3).
calculée: H 8.60 C 72.32:
trouvée: H 8.79 C 72.1 9.
3 . 5 (4s)-4-hydroxy-4-éthyl-1 -(t-butyldiphényIsilyloxy)-5-hexèn-2-
one
À la suspension du réactif de DessMartin (271 mg, 0.64 mmol) dans 20
mL de CH2C12, on ajoute lentement le composé 108 (128 mg, 0.32 mmol) dans
3 mL de CH2C12, à température ambiante. On laisse agiter 2 heures puis on
ajoute 5 mL d'une solution NaOH 1 M. On extrait plusieurs fois avec un Et20
(3x 25 mL). Les phases organiques réunies sont séchées sur Na2S04, filtrées puis concentrées. Le résidu est purifié par chromatographie éclair en utilisant comme éluant un mélange Et2O:hexanes (2:8) pour conduire au composé 110
(1 15 mg, 91 1).

IR (film). vmax (cm-'):
RMN1H (CDCl3), 6 (ppm):
R M N ' ~ C (CDC13), 6 (ppm):
396.60 g / mol
3540-3400 (OH), 3060 (CH aromatiques),
2960, 2930. 2850 (CH aliphatiques), 1725 (C=O. cétone), 1430 (CHz), 11 15 (C-0).
7.63 (4H, ml H aromatiques),
7.42 (6H, m. H aromatiques), 5.72 ( IH, dd, J = 10.7, 17.3 HZ, CHz=Cf+), 5.17 (1 H, dl J = 17.3 HZ, CHH=CH-), 5.07 (1 H, d, J = 10.7 HZ, CHH=CH-), 4.14 (2H, S, -CHPOTBDPS), 3.85 (1H, SI, OH), 2.88 (1 H, dl J = 16.5 HZ, -;;tlH-CO-), 2.55 (1 H, d, J = 16.5 HZ, -çtlH-CO-), 1.51 (ZH, ml -CH7-CH3), 1.09 (9H, s, t-butyle) et
0.85 (3H, t, J = 7.4 HZ, -CH2-m) .

TBDPSO Ss À la solution du composé 110 (170 mg, 0.43 mmol) dans 25 mL de
CH2C12, on ajoute le NI NI-bis(trirnéthylsilyI)urée (440 mg, 2.15 mrnol). Le mélange est chauffé à 60 OC pour 12 heures. On laisse la suspension revenir à température ambiante puis on filtre sur célite. Le solvant est évaporé et le résidu est purifié par chromatographie selon la technique éclair en utilisant comme éluant un mélange Et2O:hexanes (5:95) pour conduire au composé 114 (81 mg, 40%).
C27H4003Si2
Huile incolore
Rt:
RMNlH (CDC13), 6 (ppm):
468.78 g 1 mol
O. 15 (Et20: hexanes 5:95)
7.68 (4H, m, H aromatiques),
7.41 (6H. ml H aromatiques), 5.87 (1 H, dd, J = 10.7, 17.5 HZ, CHz=S;tL-), 5.09 (1H, dl J = 17.5 HZ, CBH=CH-), 5.03 (1 Hl dl J = 10.7 HZ, CHH=CH-), 4.24 (1 HI d, J = 17.8 HZ, -aH-OTBDPS), 4.18 (1 H, d, J = 17.8 HZ, -uH-OTBDPS), 2.61 (1 H, d, J = 13.9,Hz. -Cl+H-CO-), 2.54 (1 Hl d, J = 13.9.H~. -çtlH-CO-), 1.66 (2Hl m. -CH?-CH3), 1.10 (9H, s, t-butyle), 0.85 (3H, t, J = 7.4 Hz, - C H 2 - m ) et 0.00 (9H, S. TMS).

On place dans un tricol le PdC12 (4.0 mg, 0.012 mmol), le CuCl2 (62 mg,
0.360 mmol) et le AcONa (40 mg, 0.480 mmol). On y ajoute ensuite la solution
du diol 108 (48 mg, 0.12 mmol) dans de I'AcOH (1 0 mL). On fait barbotter du
CO dans la solution, qui passe initialement d'une couleur verte à une couleur
rouille. On laisse le mélange agiter ainsi pour 24 heures. Suite à cette période,
le mélange est dilué dans du benzène puis filtré sur cellulose. La phase
organique recueillie est lavée successivement avec des solutions de NaHC03
saturée et NaCl saturée. Une huile est obtenue après séchage de la phase
organique sur MgS04, filtration et évaporation. Ce résidu est ensuite
chromatographié selon la technique éclair en utilisant comme éluant un
mélange Et2O:hexanes (4:6) pour conduire au bicycle 116 (31 mg, 61%).
C25H3204Si Huile incolore
Rc
424.61 g l mol
0.41 (Et2O: hexanes 4:6)
IR (film), vmax (cm-'): 3060, 3040 (CH aromatiques), 2920, 2850
(CH aliphatiques), 1 775 (GO, lactone),

RMNiH (CDC13), 6 (ppm):
RMN13C (CDC13), 6 (ppm):
1470, 1460, 1425 (CHz), 1180, 1140, 1110 et 1065 (C-O).
7.65 (4H, ml H aromatiques),
7.40 (6H. m. H aromatiques), 4.44 (1 HI d, J = 4.9 Hz, H sur jonction de
cycle), 4.25 (1 Hl m, -=-CH2-OTBDPS), 3.78 (1 H, dd, J = 3.9, 1 1 .O HZ, -CBH- OTBDPS), 3.65 (1 H, dd, J = 3.9, 1 1 .O HZ, -CHH- OTBDPS), 2.70 (2H, m, -m-C(Et)-) , 2.32 (1 H, dd, J = 5.8, 13.7 HZ, -CH-;tLH- C O-O-), 2.01(1 H, dd, J = 9.4, 13.7 HZ, -CH-ÇtlH- CO-O-), 1.79 (2H, rn, -CH?-CHg), 1 .O5 (9H1 s, t-butyle) et 1 .O1 (3H, t, J = 7.4 Hz, CH3).

À la suspension de LiAIH4 (2.95 g, 77.75 mmol) dans de l'éther (250 mL), à O OC, on ajoute la solution de Zéthylacroleine (tech. 85%, 25.0 g, 252.50 mmol. dans 75 mL d'éther). On laisse agiter 2 keures à cette température. II est
parfois nécessaire d'ajouter à ce stadeci une petite portion de LiAIH4 (0.1 éq.) afin de compléter la réaction. On ajoute ensuite lentement de l'eau. La solution
est agitée fortement pendant 30 minutes puis filtrée sur célite. On sépare alors
les deux phases et la phase aqueuse est extraite de nouveau avec de l'éther. Les phases organiques réunies sont séchées sur Na2S04 puis filtrées. Après
évaporation du solvant, le résidu est purifié par distillation sous pression réduite pour conduire à l'alcool allylique 1 17 (1 9.14 g, 88W).
C5H10O Liquide incolore
IR (film), vmax (m-l):
RMN 'H (CDC$), 6 (ppm):
72-74 O C (50 mm de Hg) litt.137: 132 O C (760 m m de Hg)
3500-3140 (OH), 2960, 2920, 2870 (CH aliphatiques), 1650 (C=C), 1450, 1400 (CHz), 1070 et 1020 (C-O).
4.95 (1 Hl s, =CHH),
4.82 (1 Hl s, =CHH),
4.02 (2H, S, =-OH), 2.43 (1 H, SI, OH), 2.03 (2H, q, J = 7.4 Hz, CHPCH~) et 1 .O2 (3H, t, J = 7.4 HZ, CH2-CH?).

RMN I3C (CDCIB), 6 (ppm): 150.5, 107.8, 65.6, 25.5 et 11.9.
Les données spectroscopiques pour ce composé sont en accord avec celles retrouvées dans la littérature.137
À la suspension de tamis moléculaire 4A en poudre (720 mg, dans 50 mL de CHZCI~), on ajoute le L-(+)-di-iso-propyltartrate (335 mg, 1.43 mmol, dans 2 mL de CH2C12) puis I'iso-propoxyde de titane (342 IL, 1.16 mmol), à -20 C. Après 30 minutes, I'hydroperoxyde de t-butyle (4 M dans du toluène, 11.6 mL, 46.40 mmol) est ajouté goutte à goutte. Le mélange est agité pour encore 30 minutes puis l'alcool allylique 11 7 (2.00 g, 23.22 mmol) dans 5 mL CH2C12 est ajouté lentement. On laisse agiter à -20 O C pendant 15 heures puis on ajoute de l'eau (7 mL). On laisse le mélange revenir à température ambiante puis on ajoute 7 mL d'une solution NaOH 30%. La suspension est agitée fortement pendant 30 minutes puis filtrée sur célite. Le résidu est rincé abondamment avec du CHzCI2. La phase organique est séparée et la phase aqueuse est extraite plusieurs fois avec du CH2CI2. Les phases organiques réunies sont séchées sur Na2S04, filtrées puis concentrées. Le résidu est purifié par chromatographie éclair en utilisant un mélange Et20:hexanes (1 : 1) pour conduire à l'époxyde 11 8 (1.19 g, 50%). L'analyse RMN H 300 MHz de l'ester de Mosherl37 (dérivé de l'acide S-(-)s-méthoxy-a-(trif1uorométhyl)- phénylacétique) dans CDC13 indique deux doublets a 6 4.25 et 4.20 dans un
rapport correspondant à un ee de 88% pour la formation de cet isomère.
C5Hl002 Liquide incolore

Rt:
[a] *SD:
IR (film), vmax (cm-1):
RMN I H (CDCb), 6 (ppm):
O. 16 (Et2O: hexanes 1 : 1 )
3500-3300 (OH), 2960, 2920,2870 (CH
aliphatiques), 1455 (CHz), 1 O80 et 1 040
(C-O).
3.69 (2H, m, -CH?-OH), 2.86 (1 H, d, J = 4.7 HZ, XdH-0-), 2.66 (1 H, d, J = 4.7 HZ, -CHH-O-), 1.88 (1 Hl SI, OH),
1.79 (1 HI ml -çtLH-CH3), 1.57 (1 H, ml -;tLH-CH3) et 0.93 (3H, t, J = 7.5 HZ, -CHz-Q&).
RMN 1% (CDCIJ), 6 (ppm): 62.6, 60.5, 49.3, 24.3 et 8.4.
Les données pour ce composé sont en accord avec celles retrouvées dans la littérature- 38
3.1 0 (2R)-1 -(t-butyldiphénylsilyloxym6thyl)-2-éthyl-2,3-lpoxy- propane
La Et3N (2.85 mL, 20.42 mmol) est ajoutée à la solution du composé 118
(1.39 g, 13.61 mmol) dans du CH2C12 (50 mL), à O O C . On laisse agiter 10
minutes puis on additionne le DMAP (17 mg, 0.14 mmol) et le TBDPSCI (3.89
mL. 14.97 mmol). On laisse agiter encore une heure à O OC, puis 18 heures à température ambiante. Le mélange est lavé successivement avec des

solutions de NH4CI saturée, NaHC03 saturée puis NaCl saturée. La phase
organique est ensuite séchée sur MgSO4, filtrée puis concentrée. Le résidu est purifié par chromatographie éclair en utilisant un mélange Et2O:éther de pétrole
35-60 OC (3:97) pour conduire au composé 119 (3.43 g, 74%).
C21 H2802Si Huile incolore
Rt:
RMN 1H (CDC$), 6 (ppm):
340.54 g / mol
0.54 (Et20:éther de pétrole 35-60 OC 3:97)
3060, 3030 (CH aromatiques), 2950, 2920,
2850 (CH aliphatiques), 1470, 1460, 1425
(CH2) et 11 10 (C-0).
7.68 (4H, m, H aromatiques),
7.41 (6H, ml H aromatiques),
3.70 (2H, s, CH2 époxyde), 2.64 (2H, dd, J = 15.6, 4.9 HZ,-CH7-
OTBDPS), 1.81 (1 H, m. -çtLH-CH3),
1.67 (1 H, m. -çtLH-CH3),
1 .O6 (9H, s, t-butyle) et
0.91 (3H, t, J = 7.5 HZ, -CH2-CH3).
RMN 1% (CDCIJ), 6 (ppm): 135.6, 133.3, 129.6, 127.6, 65.5, 60.2,
48.9, 26.7, 24.4, 19.2 et 8.6.
Analyse élémentaire: calculée: H 8.29 C 74.07;
trouvée: H 8.51 C 74.06.

TBDPSO %OoEt O
À la suspension de NaH (60% dans l'huile, 595 mg, 14.88 mrnol) dans du DMF (75 mL), on ajoute lentement, à température ambiante, le diéthylmalonate (2.30 mL. 14.66 rnmol). La solution devient limpide lorsque tout le NaH est consommé. On ajoute alors la solution de l'époxyde 120 (2.50 g, 7.34 mmol,
dans 15 mL de DMF) et on porte à reflux pour 12 heures. On laisse le mélange revenir à température ambiante puis on ajoute une solution de HCI 10%. On agite fortement pendant 30 minutes puis on extrait avec de l'éther. Les phases organiques sont lavées avec une solution de NaHC03 saturéee, suivie d'une
solution de NaCl saturée. Après séchage des phases organiques sur MgS04, filtration et concentration, le résidu est purifié par chromatographie éclair en utilisant comme éluant un mélange Ac0Et:éther de pétrole 3540 OC (25175). On recueille le composé 121 (3.17 g, 95%), sous forme d'un mélange -1:1.2 de diastéréoisomères.
C26H3405Si
Huile incolore
R t:
IR (film), v,,, (cm-' ):
RMN 'H (CDCls), 6 (ppm):
454.64 g I mol
0.28 (Ac0Et:éther de pétrole 3540 O C 25:75)
3050 (CH aromatiques), 2950, 2920, 2850
(CH aliphatiques), 1775 (C=0, lactone), 1730 (C=O, ester), 1460. 1425 (CH2). 1150 et 11 10 (C-O).
7.65 (4H, m, H aromatiques),
7.39 (6H. m, H aromatiques), 4.24 (2H, q, J = 7.2 Hz, -çH2-CH3 de l'ester),

3.91 [3.73] (1 Hl t, J = 10.1 HZ, -CO-çtl-), 3.65 (1 H, d, J = 10.9 HZ, -MH-OTBDPS)l
3.50 (1 H, d, J = 10.9 HZ, -çtlH-OTBDPS),
2.50 (2H, d, J = 10.1 HZ, CH>-CH-CO-O-), 1.68 (2H, m, 4H7-CHJ), 1.33 (3H, t, J = 7.2 Hz, CH2-ck de l'ester), 1 -05 (9H. S. t-butyle) et 0.92 (3H. t, J = 7.4 HZ, -CHz-CH3).
RMN I3C (CDC13), 6 (ppm): 171.8 [171.4], 168.4 [168.2], 135.6 [135.5],
132.6 [l32.O], 129.9 [129.7], 727.8 [lZ7.7], 87.5 [87.5], 68.6 [66.7], 61.9 [61.4], 48.3
[47.7], 32.0 [30.7], 29.0 [28.9], 26.7 [26.6], 19.0 [19.0], 14.0 [14.0] et 7.4 r.11.
Analyse é lémentaire: calculée: H 7.54 C 68.69;
trouvée: H 7.70 C 68.67.
À la suspension de NaH (60 % dans l'huile, 147 mg, 3.65 mmol) dans du THF (50 mL), on ajoute lentement, à température ambiante. la solution du
composé 120 (1.51 g, 3.32 mrnol dans 5 mL de THF). La solution devient
limpide lorsque tout le NaH est consommé. On ajoute alors l'iodure d'éthyle (318 PL, 3.98 mmol) et on porte a reflux pour 12 heures. On laisse le mélange
revenir à température ambiante puis on ajoute une solution de NH4CI saturée.
On extrait plusieurs fois avec de l'éther. Après séchage des phases organiques

sur MgS04. filtration et concentration, le résidu est purifié par chromatographie
éclair en utilisant comme éluant un mélange Et0:éther de pétrole 35-60 O C ( : 9 ) On recueille le composé 121 (1.44 g, 90%). sous forme d'un mélange -1 :1.7 de diastéréoisomères.
C28H3805Si Huile incolore
Rt:
482.69 g 1 mol
O. 1 1 (Et2O:éther de pétrole 35-60 OC 1 :9)
IR (film), vmax (cm-'): 3050 (CH aromatiques), 2960, 2920, 2850
(CH aliphatiques), 1 770 (C=O, lactone), 1725 (C=O, ester), 1470, 1460, 1425 (CHz), 1220 et 1 1 1 O et 1090 (C-O).
RMN 1H (CDC13), 6 (ppm): 7.66 (4H, rn, H aromatiques),
7.42 (6H1 rn, H aromatiques),
4.24 [4.00] (2H, m. -CH7-CH3 de l'ester), 3.61 (2H1 m, -O-CO-CiCti2-CH3)-), 2.80 [2.60] (1 HI dl J = 13.7 HZ, -CH& OTBDPS), 2.28-1.67 (5H. m, -CH2-CJCH7-CH3)- et
CHB-OTBDPS), 1.29 (3H, t, J = 7.0 HZ, CH2-j33 de l'ester), 1.06 (9H, s, t-butyle) et
0.92 (6H, m, -CHz-Ci-l3).
RMN l3C (CDC13), 6 (ppm): 174.0 (1 74.01 , 170.4 [170.1] , 135.5 [134.7],
132.8 [132.5], 129.8 [129.7], 127.7 [127.6], 86.0 [85.6], 66.8 [66.3], 62.0 [61.9], 56.4 [%.O], 36.3, [35.5], 34.0, [32.5], 29.6 (29.31,
26.7 [26.4], 19.1 [19.1], 13.9 [13.7], 9.2 [9.0] et 7.6 [7.4].
Analyse élémentaire: calculée: H 7.94 C 69.67; trouvée: H7.90 C69.61.

TBDPSO 2;-
On porte a reflux pour 6 heures une solution du composé 121 (740 mg, 1.53 mmol) et de LiBr (670 mg, 7.70 mmol) dans du DMF non anhydre (40 mL). On laisse le mélange revenir à température ambiante puis on ajoute de la
glace. On extrait plusieurs fois avec de l'éther. Après séchage des phases organiques sur MgS04, filtration et concentration, ie résidu est purifié par
chromatographie éclair en utilisant comme éluant un mélange Et2O:éther de
pétrole 35-60 OC (1:9). On recueille le composé 122 (597 mg, 95%), sous forme d'un mélange -1 : 1.2 de diastéréoisorneres.
C25H3403Si Huile incolore
Rfi
41 0.63 g I mol
0.16 et 0.21 (Et20:éther de pétrole 35- 60 OC 1:9)
IR (film), v,,, (cm-1): 3060, 3040 (CH aromatiques), 2950, 2920,
2850 (CH aliphatiques), 1765 (C=O, lac- tone), 1470, 1460, 1425 (CHZ), 121 5 et
1 1 1 O (C-O).
RMN 'H (CDCIB), 6 (ppm): 7.66 (4H. m, H aromatiques),
7.42 (6H, m, H aromatiques), 3.68 (1 H, d, J = 10.5 HZ, CHH-OTBDPS), 3.56 (1 H, d, J = 10.5 HZ, CHH-OTBDPS), 2.81 [2.63] (1 Hl m, CH),
2.40 [2.08] (2H, m, CHPCH~), 1.92 [1.49] (2H, m. CHPCH~), 1.66 (2H. m, 4H7-CH(Et)-), 1 .O5 (9H. s, 1-butyle),

RMN l3C (CDC13), 6 (ppm):
Analyse élémentaire:
0.98 (3H. t, J = 7.4 Hz. CH2-&) et 0.89 (3H. t, J = 7.4 Hz, C H 2 a ) .
calculée: H 8.35 C 73.13;
trouvée: H 8.44 C 73.09.
3.14 Hydroxylation de la butyrolactone 122
À la solution de NaHMDS (1 .O M dans du THF, 19.0 mL, 19.0 mmol) dans
du THF (90 mL), on ajoute la solution du composé 122 (3.93 g, 9.57 mmol,
dans 20 mL de THF), à -78 OC. On laisse agiter pour une heure puis on ajoute une solution pré-refroidie à -78 O C de (+)-(8,B-dichlorocamphoryl)sulphonyI- oxaziridine (3.14 g, 10.53 mmol, dans 50 ml de THF) via cannula. La solution
est agitée pendant 4 heures puis on ajoute alors un peu d'une solution de NH4CI saturée et de l'éther. On laisse le mélange revenir à température
ambiante, on agite fortement la solution et on ajoute suffisamment d'eau pour dissoudre tous les sels. On évapore la majeure partie du THF puis on extrait
ensuite le résidu plusieurs fois à l'éther. Après séchage des phases organiques sur Na2S04, filtration et concentration, le résidu est purifié par

chromatographie éclair avec comme éluant un mélange EtzO: hexanes:CHCls (2:7:1) pour conduire majoritairement au composé 123 (2.66 g, 65%), de
même qu'une petite quantité de son épimere en C-2 124 (443 mg, 10.8%).
C25H3404Si Huile incolore
Rt:
426.63 g I mol
0.06 (Et2O:hexanes: CHCI3 2:7: 1 )
IR (film), vmx (cm-1):
RMN 'H (CDC13). 6 (ppm):
RMN I3C (CDC13), 6 (pprn):
Analyse élémentaire:
3500-3320 (OH), 3070, 3040 (CH arorna-
tiques), 2960, 2930, 2850 (CH alipha-
tiques), 1755 (C=O, lactone), 1460, 1425
(CHt), 1230 et 1 1 10 (C-0).
7.65 (4H. m, H aromatiques),
7.42 (6H, in, H aromatiques), 3.67 (1 H, d, J = 11 .O HZ, -CHH-OTBDPS), 3.54 (1 H, d, J = 11 .O HZ, -CHH-OTBDPS), 2.39 (1 H, dl J = 14.1 HZ, -CHH-C(OH)(Et)-), 2.27 (1 Hl s, OH), 2.02 (1 Hl dl J = 14.1 HZ, -CH&C(OH)(Et)-),
1.88 (2H, m, u - C H 3 ) . 1.67 (2H, m, &-CH3), 1.05 (9H, s, t-butyle), 0.96 (3H, 1, J = 7.4 Hz, CH~-çt[3) et 0.91 (3H, t, J = 7.4 HZ, C H 2 - m ) .
calculée: H 8.03 C 70.38; trouvée: H 8.31 C 70.18.

TBDPSO O
C ~ S H 3404s i Huile incolore
R f.
IR (fi lm), vmax (un-' ):
RMN 1H (CDC13), 6 (ppm):
426.63 g l mol
3500-3320 (OH), 3070, 3040 (CH aroma-
tiques), 2960, 2930, 2850 (CH alipha- tiques), 1765 (C=O, lactone), 1460, 1425 (CHz), 1230 et 11 10 (C-0).
7.67 (4H, ml H aromatiques),
7.44 (6H, ml H aromatiques), 3.94 (1 H, s, OH), 3.75 (1 H, d, J = 11 .O HZ, -CHH-OTBDPS), 3.53 (1 H, d, J = 11 .O HZ, -CH&OTBDPS), 2.27 (lHl dl 5 = 14.1 HZ, -CBH-C(OH)(Et)-), 2.20 (1 Hl d, J = 14.1 HZ, -CH&C(OH)(Et)-), 1.89 (1 H, m, mH-CH3), 1.74 (1 H, m, ÇtLH-CH3). 1-60 (2H, m, ÇH7-CH3). 1.06 (9H, s, t-butyle), 1.01 (3H, t, J = 7.5 Hz, CH2-m3) et 0.84 (3H, t, J = 7.4 HZ, C H 2 - m ) .

RMN 1% (CDC13), 6 (pprn):
Analyse élémentaire: calculée: H 8.03 C 70.38; trouvée: H 7.99 C 70.36.
À une solution du composé 123 (270 mg, 0.63 mmol) dans du THF (50
mL) à -78 OC, on ajoute lentement le DIBAL-H (1 .O M dans du toluène, 3.2 mL, 3.20 mmol). On laisse agiter à cette température pour 3 heures puis on ajoute
un peu d'une solution de NH4CI saturée. On laisse le mélange revenir à
température ambiante. Par la suite, on ajoute un peu d'une solution de HCI 10% et de l'éther, et la solution est agitée fortement pendant quelques minutes.
On effectue plusieurs extractions à l'éther et les phases organiques réunies
sont lavées avec une solution saturée de NaHC03 puis avec une solution de
NaCl saturée. Après séchage des phases organiques sur Na2S04, filtration et
concentration, le résidu est purifié par chromatographie éclair en utilisant comme éluant un mélange AcOEt:CH2C12 (595) pour conduire au lactol 128 (270 mg, 100%), sous forme d'un mélange d'isomères.
C25H3604Si Huile incolore
Rt.
428.63 g f mol
0.17 (AcOEt:CH2CI2 5:95)

IR (film), vmax (cmq):
RMN 1H (CDCI3), 6 (ppm):
3500-3280 (OH), 3060, 3040 (CH aromati-
ques), 2950, 2920, 2850 (CH alipha-
tiques), 1470, 1460, 1425 (CHz), 1 120 et 1040 (C-O).
7.68 (4H. m. H aromatiques),
7.42 (6H, rn, H aromatiques), 5.08 [4.96] (1 Hl s [d, J = 8.5 HZ], -çH-OH), 4.03 (1 H, SI. OH), 3.57 [3.46] (1 H, d, J = 10.7 HZ, W H -
OTBDPS), 3.29 [3.37] (1 H, dl J = 10.7 HZ, m H - OTBDPS),
2.14 (1 H, m, C(Et)-CBH-C(Et)),
2.03 (IH, SI, OH), 1.85-1.46 (5H, m, -CH7-CH3 et C(Et)-CHH-
C(Et))l 1.09 (1 .O61 (9H, s, t-butyle) et 0.98 [0.82] (6H, m, CH2-CHj).
RMN 1% (CDC13), 6 (ppm): 135.7 [135.6], 133.2 [132.0], 130.0 [129.6],
127.8 (1 27-61, 1 03.3 [IO1 .O], 87.6 [85.0],
83.9 [80.3], 67.9 [67.6], 41.9 [39.5], 31.2
[30.7], 28.4 [28.4], 26.7 [26.7], 19.1 [19.0], 8.7 [8.4] et 8.2 [8.0].
Analyse élémentaire: calculée: H 8.47 C 70.05;
trouvée: H 8.72 C 70.20.

A la suspension de t-BuOK (2.86 g, 24.24 mmol) dans de l'éther (400 mL), on ajoute le bromure de méthyltriphénylphosphonium sec (8.61 g, 24-10 mmol), a température ambiante. On laisse agiter 2 heures puis on ajoute le lactol 128
(2.07 g, 4.85 mmol) en solution dans de l'éther (50 mL). Le mélange est porté à
reflux pour 15 heures. On ajoute alors un peu d'eau et on extrait plusieurs fois avec un mélange Et20:éther de pétrole 35-60 O C (2:l). Les phases organiques réunies sont séchées sur Na2S04, filtrées puis concentrées. Le résidu est
purifié par chromatographie éclair en utilisant comme éluant un mélange
AcOEt:CH2C12 (5:95) pour conduire au diol 129 (1 -27 g, 61 %).
C26H3803Si Huile incolore
R f i
426.67 g / mol
0.12 (AcOEt:CH2C12 5:95)
[a] $5: -2.77 (C 0.62, CHC13)
IR (film), v,,, (un-1):
RMN 1H (CDC13), 6 (ppm):
3500-3200 (OH), 3060, 3040 (CH aromati-
ques), 2960, 2920, 2850 (CH aliphati- ques), 1470, 1460, 1425 (CHâ), 1 1 1 O et 1 O80 (C-O).
7.65 (4H1 ml H aromatiques),
7.41 (6H, m, H aromatiques), 5.68 (lH, dd, J = 10.6, 17.2 HZ, H2C=C&), 5.22 (IH, d, J = 17.2 Hz, HHC=CH-), 4.89 (1 H, dl J = 10.6 Hz, HHC=CH-), 4.32 (1 H, SI, OH),

3.87 (IH, dl J = 10.0 HZ -CHH-OTBDPS),
3.46 (1 H, d, J = 10.0 HZ, -CH&OTBDPS), 2.99 (AH, SI, OH), 1 -86 (1 H, d, J = 15.1 HZ, C(Et)-CHH-C(Et)),
1.65 (1 H, d, J = 15.1 HZ, C(Et)-CHH-C(Et)), 1.53 (2H, m, -CH?-CH3), 1.39 (2H. m. -CH2-CH3), 1.09 (9H, s, t-butyle),
0.83 (3H, t, J = 7.4 Hz, -CH~-çti3) et 0.82 (3H. t, J = 7.4 HZ, -CH2-çti3).
RMN I3C (CDC13). 6 (ppm): 144.4, 135.5, 132.9, 129.7. 127.6, 112.3,
75.4, 67.3, 65.7, 42.6, 36.2, 32.5, 26.8,
19.1, 7.6 et 7.3.
Analyse élémentaire: calculée: H 8.98 C 73.19;
trouvée: H 9.27 C 73.12.
On place dans un tricol le PdCl2 (41 mg, 0.23 mmol), le CuC12 (1.20 g, 7.04 mmol) et le AcONa (770 mg, 9.39 mmol). On y ajoute ensuite la solution du diol 129 (1.00 g, 2.34 mmol) dans de I'AcOH (40 mL). On fait barbotter du
CO dans la solution, qui passe initialement d'une couleur verte à une couleur rouille. On laisse le mélange agiter ainsi pour 60 heures. Suite à cette période,
le mélange est dilué dans du benzène puis filtré sur cellulose. La phase

organique recueillie est lavée successivement avec des solutions de NaHCO3
saturée et NaCl saturée. Une huile est obtenue après séchage de la phase organique sur MgS04. filtration et évaporation. Ce résidu est ensuite chromatographié selon la technique éclair en utilisant comme éluant un mélange Et20:éther de pétrole 35-60 O C (614) pour conduire au bicycle 133 (953 mg, 90%).
C27H3604Si Solide blanc
R f
452.67 g I mol
0.40 (Et2O:éther de pétrole 35-60 O C 6:4)
P-f.: 75-76 O C (hexanes)
IR (KR), vmô, (cm-' ): 3060, 3040 (CH aromatiques), 2960, 2930,
2850 (CH aliphatiques), 1780 (C=O), 1460,
1430 (CHz), 1220, 1 1 10 et 1055 (C-O).
RMN (CDCl3). 6 (ppm): 7.66 (4H, m, H aromatiques),
7.41 (6H, m, H aromatiques),
4.42 (1 HI m, H sur jonction de cycle),
3.59 (1 H, d, J = 10.4 HZ, -CHH-OTBDPS),
3.46 (1 H, d, J = 70.4 HZ, -CH&OTBDPS), 2.66 (2H, S, -CH-CH2-CO-),
2.24 (2H, S, -C(Et)-CH,-C(Et)-),
1-68 (2H. m. -C(CH2-OTBDPS)-CH2-CH3), 1-56 (ZH, q, J = 7.6 HZ, -CO-O-C(CH2-
cH3)-)1 1 -07 (9H, s, t-butyle), 0.98 (3H, t, J = 7.4 HZ, -C(CH2-0TBDPS)- CH2-çt13) et
0.83 (3H, t, J = 7.6 HZ, -CO-O-C(CH2-
Çt13)-1.

RMN 1% (CDC13), 6 (ppm):
Analyse élémentaire: calculée: H 8.02 C 70.64;
trouvée: H 8.39 C 70-35.
TBDPSO OH
A la solution du composé 124 (270 mg, 0.63 mmol) dans du THF (50 mL)
à -78 OC, on ajoute lentement le DIBAL-H (1.0 M dans du toluène, 3.2 mL, 3.20
mmol). On laisse agiter à cette température pour 3 heures puis on ajoute un peu d'une solution de NH4CI saturée. On laisse le mélange revenir à
température ambiante. Par la suite, on ajoute un peu d'une solution de HCI
10% et de I'éther et la solution est agitée fortement pendant quelques minutes.
On effectue plusieurs extractions à I'éther et les phases organiques réunies
sont lavées avec une solution saturée de NaHC03 puis avec une solution de
NaCl saturée. Après séchage des phases organiques sur Na2S04, filtration et concentration, le résidu est purifié par chromatographie éclair en utilisant
comme éluant un mélange Et20: éther de pétrole 35-60 O C (3:7) pour conduire
au lactol 134 (260 mg, 96%)' sous forme d'un mélange d'isomères.
C25H3604si Huile incolore
R f i
428.63 g ! mol
0.26 (Et20: éther de pétrole 35-60 OC 3 7 )

IR (film), v,, (an-' ):
RMN 1H (CDC13). 6 (ppm):
3500-3240 (OH), 3050, (CH aromatiques),
2960, 2930, 2850 (CH aliphatiques), 1 460, 1430 (CHz), 1 160, 1 1 10, 1070, 1035 et
1 O00 (C-O).
7.71 (4H. rn, H aromatiques), 7.43 (6H, m, H aromatiques), 5.20 [4.90] (1 H, d, J = 2.4 Hz [ml, -=OH),
4.13 [3.40] (1 H, dl J = 12.2 HZ, M H -
OTBDPS), 3.64 [3.33] (1 Hl dl J = 10.5 HZ, m- OTBDPS), 2.61 (2H, SI, 2x OH),
2.16 (1 Hl ml C(Et)-CHH-C(Et)),
1.98 (1 H, m, C(Et)-CHH-C(Et)), 1 -74 (1 H, ml -CjjH-CH3), 1.61 (1 Hl ml -CHH-CH3).
1.49 (1 Hl m. -CMH-CH3),
1.37 (1 Hl m l -CuH-CH3), 1 .O9 [1.07] (9H, s, t-butyle), 0.80 [1 .O61 (3H. t, J = 7.4 Hz, CH2-&) et
0.72 [1.02] (3H, t, J = 7.4 HZ, CH2-CH3).
RMN 1% (CDCI3). 6 (ppm): 135.6 [135.5], 131 -9 [131 -71, 130.0 [129.9],
127.8 [127.7], 104.4 [101.3], 87.3 [82.6], 82.1 [79.0], 69.6 [68.5], 43.0 [41.4], 33.9
[SI .3], 28.2 [26.9], 26.6 [26.6], 18.1 [15.5], 8.8 (8.51 et 8.3 [7.8].
Analyse élémentaire: calculée: H 8.47 C 70.05;
trouvée: H 8.67 C 70.38.

À la suspension de t-BuOK (665 mg, 5.60 mmol) dans de l'éther (90 mL),
on ajoute le CH3PPh3Br sec (2.0 g, 5.60 mmol), à température ambiante. On laisse agiter 2 heures puis on ajoute le lactol 134 (240 mg, 0.56 mmol) en solution dans de l'éther (50 mL). Le mélange est porté à reflux pour 15 heures. On ajoute alors un peu d'eau et on extrait plusieurs fois avec un mélange
Et2O:éther de pétrole 35-60 O C (2:l). Les phases organiques réunies sont séchées sur Na2S04, filtrées puis concentrées. Le résidu est purifié par chromatographie éclair en utilisant comme éluant un mélange AcOEt:CH2C12
(3197) pour conduire au diol 135 (71 mg, 30%).
426.67 g / mol
O. 15 (AcOEt:CH2C12 3:97)
IR (film), v,, (cm-1 ):
RMN 'H (CDCI3), 6 (ppm):
3500-3260 (OH), 3050 (CH aromatiques),
2950, 2920, 2850 (CH aliphatiques), 1460, 1430 (CH2), 11 10 et 1080 (C-O).
7.66 (4H, m, H aromatiques),
7.42 (6H, m, H aromatiques), 5.92 (1 H, dd, J = 10.5, 17.1 HZ, H2C=çtL-),
5.38 (1 HI dd, 5 = 1.9, 17.1 Hz, HHC=CH-),
5.12 (1H, dd, J = 1.9, 10.5 HZ, HHC=CH-), 4.37 (1 HI SI, OH),
3.48 (2H, m -CHPOTBDPS),

2.89 (1 Hl SI, OH), 1.86-1 -38 (6H1 rn, 3x -CH2-), 1 .O9 (9H, s, t-butyle),
0.84 (3H. t, J = 7.4 Hz, -CH?=) et
0.71 (3H, t, J = 7.4 HZ, -CH2-m) .
RMN 1% (CDC13), 6 (ppm): 144.6, 135.5, 132.6, 129.8, 127.7, 112.4,
76.5, 75.2, 69.1, 43.0, 36.2, 29.8, 26.8, 19.2, 8.0 et 7.3.
Analyse élémentaire: calculée: H 8.98 C 73.19; trouvée: H 9.04 C 73.12.
TBDPSO C
H
On place dans un tricol le PdC12 (41 mg, 0.23 mmol), le CuCIz (1.20 g,
7.04 mmol) et le AcONa (770 mg, 9.39 mmol). On y ajoute ensuite la solution
du di01 135 (1.00 g, 2.34 mmol) dans de I'AcOH (40 mL). On fait barbotter du
CO dans la solution, qui passe initialement d'une couleur verte à une couleur rouille. On laisse le mélange agiter ainsi pour 60 heures. Suite à cette période,
le mélange est dilué dans du benzène puis filtré sur cellulose. La phase
organique recueillie est lavée successivement avec des solutions de NaHC03 saturée et NaCl saturée. Une huile est obtenue après séchage de la phase organique sur MgS04, filtration et évaporation. Ce résidu est ensuite chromatographié selon la technique éclair en utilisant comme éluant un mélange Et2O:éther de pétrole 35-60 OC (6:4) pour conduire au bicycle 136 (953 mg, 90%).

C27H3604S i Huile incolore
Rr-
RMN 1H (CDC13), 6 (ppm):
RMN 1% (CDCIJ), 6 (ppm):
452.67 g 1 mol
0.35 (Et20:éther de pétrole 35-60 O C 6:4)
3050, 3030 (CH aromatiques), 2950, 2930,
2850 (CH aliphatiques), 1775 (C=O). 1470. 1425 (CH2). 1180, 1110 et 1055 (C-O).
7.65 (4H, m, H aromatiques),
7.40 (6H, rn, H aromatiques), 4.38 (IH, dl J = 4.4 Hz, H sur jonction de
cycle), 3.55 (2H, S, -Cb-OTBDPS), 2.64 (2H, m, -Ci+- du groupement Et sur jonction de cycle), 2.52 (IH, d, J = 14.6 Hz, H sur Cô), 1 -93 (1 H, d, J = 14.6 Hz. H sur Cô), 1.82-1.59 (4H. m, CO-CH?, - C H A H 3 sur
c-7 ) , 1 .O6 (9H, s, t-butyle), 1-01 (3H, t, J = 7.4 HZ, -C(CH2-0TBDPS)-
CH2-CH3) et 0.84 (3H, t, J = 7.4 HZ, -CO-O-C(CH2-
Çt13)-1-

TBDPSO OH
À une solution du composé 122 (256 mg, 0.62 mmol) dans du THF (50 mL) à -78 OC. on ajoute lentement le DIBAL-H (1 .O M dans du toluène, 1.25 mL, 1.25 mmol). On laisse agiter à cette température pour 3 heures puis on ajoute
un peu d'une solution de NH4CI saturée. On laisse le mélange revenir à température ambiante. Par la suite. on ajoute un peu d'une solution de HCI
10% et de l'éther et la solution est agitée fortement pendant quelques minutes. On effectue plusieurs extractions à t'éther et les phases organiques réunies sont lavées avec une solution saturée de NaHC03 puis avec une solution de
NaCl saturée. Après séchage des phases organiques sur Na2S04, filtration et concentration, le résidu est purifié par chromatographie éclair en utilisant comme éluant un mélange Et2O:éther de pétrole 35-60 OC (1 5:85) pour conduire au lactol 137 (205 mg, 80%). sous forme d'un mélange d'isomères.
C25H3603Si
Huile incolore
Rt
41 2.64 g I mol
0.1 7 et 0.32 (Et20:éthe: de pétrole 35-60 O C l5:85)
IR (film), vmax (an-' ): 3500-3260 (OH), 3060. 3040 (CH aroma-
tiques), 2960. 2930, 2845 (CH aliphati-
ques), 1460. 1430 (CHz), 1 1 10 et 1 O00 (C-
o 1-
RMN 'H (CDC13). 6 (ppm): 7.71 (4H, m, H aromatiques),
7.43 (6H, ml H aromatiques), 5.20 [4.90] (1 H, dl J = 2.4 Hz [2d, J i = 11 -9 HZ. J2 = 13.7 HZ], -Ct+OH),

RMN 13C (CDC13), 6 (ppm):
Analyse élémentaire:
4.1 3 [3.40] (1 Hl dl J = 12.2 HZ, ÇtiH- OTBDPS), 3.64 [3.33] (1 Hl dl J = 10.5 HZ, ÇtlH- OTBDPS), 2.61 (ZH, SI, 2x OH), 2- 16 (1 Hl M, C(E1)-CHH-C(Et)), 1.98 (1 Hl m. C(Et)-CHHC(Et)), 1.74 (1 H, m. -CbH-CH3), 1-61 (IH, m, -C&l-CH3), 1.49 (1 HI m, ÇHH-CH3). 1.37 (1 Hl rn, -Cj-JH-CH3), 1 .O9 [1.07] (9H, s, t-butyle), 0.80 [1 .CE] (3H, t, J = 7.4 Hz, C H 2 - W ) et 0.72 [1.02] (3H, t , J = 7.4 HZ, C Hz-C H3).
Étant donné que le composé 137 est un
mélange complexe d'isomères, le RMN I3C donne lieu à un spectre très compli- qué dont il s'avère inutile de dresser une liste exhaustive.
calculée: H 8.79 C 72.77; trouvée: H 9.00 C 72.82.

3.22 (SR)-3,5-diéthyl-5-(f-butyldiph~nylsilyloxyméthyl)-2,3- di hydrofurane
TBDPSO
138
À une solution du lactol 138 (184 mg, 0.45 mmol) dans 10 mL de CH2C12, on ajoute la Et3N (240 PL, 1.71 mmol) puis le MsCl (45 PL, 0.58 mmol), à -20
OC. On laisse le mélange revenir à température ambiante puis on laisse agiter pour 4 heures. Le mélange est ensuite porté à reflux pour 12 heures. On laisse le mélange revenir à température ambiante puis on lave la phase aqueuse successivement avec 10 mL d'une solution HC1 IO%, I O mL d'une solution de
NaHC03 saturéee et de 10 mL d'une solution de NaCl saturée. Après séchage des phases organiques sur MgS04, filtration et concentration, le résidu est purifié par chromatographie éclair en utilisant comme éluant un mélange
Et2O:éther de pétrole 3560 OC (2:98) pour conduire à l'alcène 137 (80 mg, 45%).
C25H3402Si
Huile incolore
Rt:
IR (film), vmax (cm-' ):
RMN 1i-i (CDC13), 6 (ppm):
394.63 g I mol
0.44 (Et2O:éther de pétrole 35-60 OC 2:98)
3060. 3040 (CH aromatiques), 2960, 2920,
2845 (CH aliphatiques), 1665 (C=C), 1460, 1425 (CH2) et 1 1 10 (C-0).
7.70 (4H, m, H aromatiques),
7.41 (6H, ml H aromatiques), 5.87 (1 Hl S, -O-CH=C(Et)-), 3.60 (2H, S, C&-OTBDPS), 2.50 (1 Hl dl J = 14.2 HZ, =C(Et)-CBH),
2.32 (1 Hl dl J = 14.2 Hz, =C(Et)-CHH),

RMN 13C (CDCJ3). 6 (ppm):
Masse exacte m/e:
(IE, 70 eV)
SM (IE, 70 eV)
2.00 (2H, q, J = 7.4 HZ, G(CH2-OTBDPS)-
CH2-CH31, 1.73 (2H. q, J = 7.4 HZ, =C-C&-CHj). 1 .O8 (9H. S. t-butyle), 1.02 (3H, t, J = 7.4 HZ, G(CH2-0TBDPS)- C H 2 - m ) et 0.91 (3H, t, J = 7.4 HZ, =C-CH2-Cfl3).
pour C21 H2502Si (M-C4Hg)+, calculée: 337.1624; trouvée: 337.161 6.

BIBLIOGRAPHIE
de Silva, E. D.; Scheuer, P.J. Tetrahedron Lett, 1980, 21, 161 1.
Scheuer, P. J. Chemistry of Marine Natural Products, Academic Press, 1973.
Manne Natural Products, P. J. Scheuer Éd., Academic Press, 1978, Vol.
1-5. Bioorganic Manne Chemistry, Scheuer, P. J. Éd., Springer-Verlag, 1987, Vol. 7.
Bioorganic Manne Chemistry, Scheuer, P. J. Éd., Springer-Verlag, 1 988, Vol. 2.
Bioorganic Manne Chemistv, Scheuer, P. J. Éd., Springer-Verlag, 1989,
Vol. 3. Bioorganic Manne Chemistry, Scheuer, P. J. Éd., Springer-Verlag, I W I , Vol. 4,.
Bioorganic Manne Chemistry, Scheuer, P. J. Éd., Springer-Verlag, 1992, Vol. 5.
Bioorganic Manne Chemistry, Scheuer, P. J. Éd., S pringer-Verlag, 1992,
Vol. 6. Faulkner, D. J. Nat. Prod. Rep. 1984, 7, 251. Faulkner, D. J. Nat. Pmd. Rep. 1984, 7, 551.
Faulkner, D. J. Nat Prod. Rep. 1986, 3, 1.
Faulkner, D. J. Nat Prod. Rep. 1987, 4, 539. Faulkner, D. J. Naf. Pmd. Rep. 1988, 5, 613.
Faulkner, D. J. Nat. Prod. Rep. 1990, 7, 269. Faulkner, D. J. Naf. Prod. Rep. 1991, 8, 97. Faulkner, D. J. Nat. Prod. Rep. 1992, 9, 323. Faulkner, D. J. Nat. Prod. Rep. 1993, 10, 497. Faulkner, D. J. Nat. Prod. Rep. 1994, 11, 355.

Faulkner, D. J. Nat. Pmd. Rep. 1995, 12, 223. Faulkner, D. J. Nat. Pmd. Rep. 1996, 13, 75. Faulkner, D. J. Nat. Prod. Rep. 1997, 14, 259. a) Yasumoto, T.; Murata, M. Chem. Rev., 1993, 93, 1897; b) Yokoyama, A; Murata, M.; Oshima, Y.; lwashita, T.; Yasumoto, T. J. Biochem., 1988, 104, 184. a) Armstrong, R. W.; Beau, J.M.; Cheon, S. H.; Christ, W. J.; Fujioka, H.; Ham, W.-H., Hawkins, L. D.; Jin, H.; Kang, S. if.; Kishi, Y., Martinelli. M. J.; McWorther, W. W., Jr.; Mizuno, M.; Nakata, M.; Stutz, A E.; Talamas, F. X.; Taniguchi, M.; Tini, J. A; Ueda, K.; Uenishi, J.; White, J. B.; Yonaga, M. J.
Am. Chem. Soc., 1989, 7 11, 7525; b) Armstrong, R. W.; Beau, J.-M.; Cheon, S. H.; Christ, W. J.; Fujioka, H.; Ham, W.-H., Hawkins, L. D.; Jin, H.; Kang, S. H.; Kishi, Y.; Martinelli, M. J.; McWorther, W. W. Jr.; Mizuno,
M.; Nakata, M.; Stutz, A E.; Talamas, F. X.; Taniguchi, M.; Tini, J. A; Ueda, K.; Uenishi, J.; White, J. B.; Yonaga, M. J. Am. Chem. Soc., 1989: 111, 7530; c) Suh, E. M.; Kishi, Y. J. Am. Chem. Soc., 1994, 116, 11205;
d) Kishi, Y. Chem. Scr., 1987, 27, 573; e) Kishi, Y. Pure & Appl. Chem., 1989, 61, 313. Nicolaou, K. C.; van Delft, F.; Ohshima, T.; Vourloumis, D.; Xu, J.;
Hosokawa, S .; Pfefferkom, J.; Kim, S.; Li, T. Angew. Chem. lnt. Ed. Engl., 1997, 36, 2520.
Borman, S. C&EN, 1997, November 24 issue, 64.
a) Gribble, G. W. Acc. Chem. Res., 1998, 31, 141: b) Harper, D. B.: O'Hagan, D. Nat Prod. Rep., 1994. 11, 123: c) Gribble, G. W. J. Chem. Ed., 1994, 71, 907; d) Gribble, G. W. J. Nat. Prod., 1992, 55, 1353; e) Fennical, W. Science, 1982, 215, 923
a) Jung, M. E.; Parker, M. H. J. Org. Chem., 1997, 62, 7094; b) Fuller, R. W.; Cardellina II, J. H.; Kato, Y.; Brinen, L. S.; Clardy, J.; Snader, K. M.; Boyd, M. R. J. Med. Chem., 1992,35, 3007. a) Kato, Y.; Fusetani, N.; Matsunaga, S.; Hashirnoto, K.; Fujita, S.; Furuya, T. J. Am. Chem. Soc., 1986, 708, 2780; b) Kato, Y.; Fusetani, N.; Matsunaga, S.; Hashimoto, K.; Koseki, K. J. Org. Chem., 1988, 53, 3930;
c) Matsunaga, S.; Fujiki, H.; Sakata, D. Tetrahedron, 1991, 47, 2999. a) Figadere, B. Acc. Chem. Res., 1995, 28, 359; b) Hoppe, R.; Scharf H.-
D. Synthesis, 1995, 1447; c) Rupprecht, J. K.; Hui, Y.-H.; McLaughlin, J. L. J. Nat. Prod., 1990, 53, 237.

a) Elliott, M. C. Cont. Org. Synth., 1997, 4, 238; b) Elliott, M. C. Cont. Org.
Synth., 1996, 3, 229; c) Harmange, J.-C.; Figadère, B. Tetrahedron: Asymmefry, 1993, 4, 171 1; d) Cardillo, G.; Orena, M. Tetrahedron, 1990, 46, 3408; e) Boivin, T. L. B. Tefrahedron, 1987, 43, 3362. Baldwin, J. E. J. Chem. Soc. Chern. Commun., 1976, 734. Abe, M.; Kiyota, H.; Adachi, M.; Oritani, T. Synlett, 1996, 777. Aldabbagh, F.; Bowman, W. R. Cont. Org. Synth., 1997, 4, 261. Burke, S. D.; Jung, K. W. Tetrahedron Letf., 1994, 35, 5837.
a) Matsuo, K; Tanaka, M.; Sakai., K; Suernune, H. Tetrahedron:
Asymmefry, 1997, 8, 3089; b) Estermann, H.; Prasad, K.; Shapiro, M. J.; Bolsterli, J. J.; Walkinshaw, M. D. Tetrahedron Le&, 1990, 31, 445; c) Naemura, K.; Takahashi, N.; Chikamatsu, H. Chem. Lett., 1988, 1717. Hoye, T. R.; Ye, Y. J. Am. Chem. Soc., 1996, 118, 1801. Koert, U.; Stein, M.; Wagner, H. Eur. J. Chem., 1997, 3, 1170. Jung, M. E.; D'Arnica, D. E. J. Am. Chem. Soc., 1997, l I 9 , 12150.
Semmelhack, M. F.; Zhang, N. J. Org. Chem., 1989, 54, 4483 et autres références citées. Malleron, J.-L.; Fiaud, J - C ; Legros, J.-Y. Handbook of Palladium- Catalyzed Organic Reactions: Synthetic Aspects and Catalytic Cycles, Academic Press, San Diego, 1997. Tsuji, J. Palladium Reagents and Catalysts: innovations in Organic Synthesis, Wiley & Sons, Chichester, 1995. Trost, B. M. Homogeneous Transition Metal Catalyzed Reactions, Moser, W. R.; Slocum, D. W. Éd., ACS, Washington, 1992, 463.
Collman, J. P.; Hegedus, L. S.; Norton, J. R.; Finke, R. G. Pnnciples and Applications of Organotransition Meta1 Chemistry, University Science Books, Mill Valley, 1987. Heck, R. F. Palladium Reagents in Organic Synthesis, Academic Press, London, 1985. Colquhoun, H. M.; Holton, J.; Thompson, D. J., Twigg, M. V. New Pa th wa ys for O rga nic Synthesis: Practical Applica fions of Transition Metals, Plenum Press, New York, 1984. Trost , B. M. Comprehensive Organometallic Chemistfy, Wilkinson, G. É d., Pergamon Press, Oxford, 1982, Vol. 8, 799. a) Semmelhack, M. F.; Bodurow, C. J. Am. Chem. Soc., 1984, 706, 1496; b) Semmelhack, M. F.; Kim, C.; Yhang, N.; Bodurow, C.; Sanner, M.;

Dobler, W.; Meier, M. Pure & Appl. Chem., 1990, 62, 2035 et références citées.
a) Kraus, G. A; Li, J.; Gordon, M. S.; Jensen, J. H. J. Org. Chem., 1995, 60, 1154; b) Kraus, G. A; Li, J.; Gordon, M. S.; Jensen, J. H. J. Am. Chem. Soc., 1993, 115, 5859; c) Graaa, T.; Jager, V. Synletf, 1992, 191 ; d) Graua, T.; Jager, V. Synthesis, 1994, 1359; e) Semmelhack, M. F.; Epa, W. R.; Cheung, A W.-H.; Gu, Y.; Kim, C.; Yhang, N.; Lew, W. J. Am. Chem. Soc., 1994, 116, 7455. Tamaru, Y.; Kobayashi, T.; Kawamura, S.-i.; Ochiai, H.; Hojo, M.; Yoshida, Z.-i. Tetrahedron Lett., 1985, 26, 3207. Tamaru, Y.; Yoshida. 2.4. J. OQ. Met. Chem., 1987, 334, 21 3. a) Tamaru, Y.; Kimura, M. Synlett, 1997, 749; b) Tamani, Y.; Hojo, M.;
Yoshida, Z.4. J. Org. Chem., 1988, 53, 5731; c) Tamaru, Y.; Kobayashi,
T.; Kawamura, S.-i.; Ochiai, H.; Yoshida, Z-i. Tetrahedron Lett., 1985, 26, 4479. Tamaru, Y.; Higashirnura, H.; Naka, K.; Hojo, M.; Yoshida, 2. Angew. Chem. Int. Ed. Engl., 1985, 24, 1045. Trost, B. M. Angew. Chem. Int. Ed. Engl., 1995, 34, 259. Corey, E. J.; Cheng, X.M. The Logic of Chernical Synthesis, John Wiley & Sons, New York, 1989. Trost, B. M. Science, 1985, 227, 908.
Nicolaou, K. C.; Sorensen E. J. Classics in Total Synthesis, VCH, New York, 1996. Wender, P. A; Handy, S.; Wright, D. L. Chem. Ind., 1997, 765. Nicolaou, K. C.; Webber, S. E. J. Am. Chem. Soc., 1984, 106, 5734.
Suzuki, T.; Koisumi, K.; Suzuki, M.; Kurosawa, E. Chem. Lett., 1983, 1643. Barrow, R. A.; Capon, R. Aust. J. Chem., 1980, 33, 891. Patil, A D.; Freyer, A J.; Bean, M. F.; Carte, 8. K.; Westley, J. W.; Johnson, R. K.; Lahouratate, P. Tetrahedron, 1996, 52, 377. a) Erickson, K. L. in Marine Natural Pmducts , P.J. Scheuer Éd., Academic Press, 1978, Vol. 3, 131; b) Obota, Y.; Fukushi, S. J. Agr. Chem. Soc. Japan, 1953, 27, 331: c) Irie, T.; Suzuki, M.; Masamune, T. Tetrahedron Leff., 1965, 1091 ; d) Anorbe, B.; Martin, V. S.; Palazon, J. M.; Trujillo, J.M. Tetrahedron Lett., 1986, 27, 4991; e) Tonn, C. E.; Palazon, J. M.; Ruiz-Pérez, C.; Rodriguez, M. L.; Martin, V. S. Tetrahedron

Leff., 1988, 29, 31 49; f) Fukuzawa, A; Aye, M.; Tkaya, Y.; Fukui, H.; Masamune, T.; Murai, A Tetrahedron Lett, 1989, 30, 3665.
Brown, M. J.; Harrison, T.; Overman, L. E. J. Am. Chem. Soc., 1991, 113, 5378. Osumi, K.; Sugimura H.; Tetrahedron Left., 1995, 36, 5789. Martin, T.; Soler, M. A; Betancort, J. M.; Martin, V. S. J. Org. Chem., 1997, 62, 1570.
Lee, E.; Yoo, S.-K.; Cho, Y.S.; Cheon, H.S.; Chong, Y. H. Tetrahedron
Lett., 1997, 38, 7757. Gringore, O. H.; Rouessac, F. P. Org. Synth., 1985, 63, 121.
Taniguchi, M.; Koga, K.; Yamada. S. Tetrahedron, 1974, 30, 3547. Eguche, C.; Kakuta, A Bull. Chern. Soc. Jpn., 1974, 47, 1704. Barrett, A G. M. in Comprehensive Organic Chemistw B. M. Trost. 1. Fleming Éd, Pergamon Press, Oxford, 1991, Vol. 8, 235.
Ravid, U.; Silverstein, R. M.; Smith, L. R. Tetrahedron, 1978, 34, 1449.
a) Brown, H. C.; Heim, P.; Yoon, N. M. J. Org. Chem., 1972, 37, 2942; b) Brown, H. C.; Heim, P.; Yoon, N. M. J. Am. Chem. Soc., 1970, 92, 1637.
a) Kunz, H.; Waldrnann, H. in Comprehensive Organic Chernistry, B.M. Trost, 1. Fleming Éd, Pergamon Press, Oxford, 1991, Vol. 6, 631 ; b) van Look, G. Silylating Agents, Fluka Chemie AG, Buchs, 1988; c) Nelson, T. D.; Crouch, R. O.; Synthesis, 1996, 1031.
Hanessian, S.; Lavallée, P. Can. J. Chem., 1975, 53, 2975.
a) Hanessian, S.; Murray, P. J. Tetrahedron, 1987, 43, 5055; b)
Hanessian, S.; Sahoo, S. P.; Murray, P. J. Tetrahedron Lett., 1985, 26,
5631.
Sato, M.; Ohuchi, H.; Abe, Y.; Kanedo, C. Tetrahedron: Asymmetry,
1992, 3, 313.
Jones, B. in Comprehensive Organic Chemisfw, B.M. Trost, 1. Fleming Éd, Pergamon Press, Oxford, 1991, Vol. 7, 151.
a) Vedejs, E.; Engler, D. A; Telschow, J. E. J. Org. Chem., 1978, 43, 188; b) Vedejs, E. J. Am. Chem. Soc., 1974, 96, 5944. Vedejs, E.; Larsen, S. Org. Synth., 1984, 64, 127. a) Davis, F. A; Lamendola, J. Jr.; Nadir, U.; Kluger, E. W.; Sedergran, T. C.; Panunto, T. W.; Billmers, R.; Jenkins, R. Jr.; Turchi, 1. J.; Watson, W. H.;

Chen, J. S.; Kimura, M. J. Am. Chem. Soc., 1980, 102, 2000; b) Davis, F.
A; Nadir, U. K; Kluger, E. W. J. Chem. Soc. Chem. Commun., 1977, 25. a) Davis, F. A.; Chattopadhyay, S.; Towson, J. C.; Lal. S.; Reddy, T. J. Org. Chem., 1988, 53, 2087; b) Vishwakama, 1. C.; Stringer, O. D.; Davis, F. A Org. Synth., 1988, 66, 203; Davis, F. A; Vishwakarma, L. C.; Billmen, J. M.; Finn, J. J. Org. Chem., 1984, 49, 3241. Mitsunobu, O. Synthesis, 1981, 13, 807.
a) Hughes, D. L. in Organic Reactions, L. A Paquette Éd, Wley & Sons,
New York, 1992, Vol. 42, 335, b) Hughes, D. L. Org. Prep. Proc. M.,
1996, 28, 127. Bhattacharjee, S. S.; Schwartz, J. A; Perlin, A. S. Carbohydr. Res., 1975, 42, 259. Roy, R.; Rey, A W. Can. J. Chem., 1991, 69.62.
Fitjer, L.; Quabeck, U. Synth. Commun., 1985, 15, 855. Moriarty, R. M.; Kim. J.; Brumer, H. III Tetrahedron Le#., 1995, 36, 51.
Radu, 1.4. Synthèse énantiosélective de la (+)-muscarine et d'un 1.3 anti-
di01 fonctionnalisé, précurseur d'un tétrahydrofurane naturel. Étude sur
la perhydrolyse des époxydes, préparation et structure de nouveaux 1,2,4-trioxanes, Université Laval, 1997. Saito, S.; Hasegawa, T.; Inaba, M.; Nishida, R.; Fujii, T.; Nomizu, S.;
Moriwake, T. Chem. Lett, 1984, 1389.
a) Banfi, 1.; Cascio, G.; Guanti, G.; Manghisi, E.; Narisano, E.; Riva, R. Tetrahedron, 1994, 50, 11967; b) Wess, G.; Klesser, E. B.; Bartmann. W.; Beck, G.; Bergmann, A; Jendralla, H.; Bock, K.; Holzstein, G.; Kleine, H.;
Schniener, M. Tetrahedron Lett, 1990, 31, 2545; c) Guanti, G.; Banfi, L.; Narisano, E. Tetrahedron Lett., 1989, 30, 5507. a) Nahm, S.; Weinreb, S. M. Tetrahedmn Leff., 1981, 22, 3815; b)
Basha, A.; Lipton, M.; Weinreb, S. M. Tetrahedmn Lefi., lS77, 48, 41 71. Evans, D. A.; Chapman, K. T.; Carreira, E. M.; J. Am. Chem. Soc., 1988,
7 10, 3560.
Evans, D. A; Gauchet-Prunet, J. A; Carreira, E. M.; Charette, A B. J. Org. Chem., 1991, 56, 741. a) Khathawala, F. G.; Prager, B.; Prasad, K.; Repic, O.; Shapiro, M. J.; Stabler, R. S.; Wilder, L. Helv. Chim. Acta, 1986, 69, 803; b) Chen, K.-M.;

Hardmann, G. E.; Prasad, K.; Repic, O.; Shapiro, M. J. Tetrahedron Lett.,
1987, 28, 155.
Corey, E. J.; Ruden, R. A Tetrahedron Lett., 1973, 1495.
a) Kuwajima, i.; Nakamura. E.; Shimizu, M. J. Am. Chem. Soc., 1982, 104, 1025; b) Paquette, L. A; Doherty, A M.; Rayner, C. M. J. Am. Chem. Soc., 1992, 714, 3910. Mancuso, A J.; Huang, SA.; Swem, D. J. Org. Chem., 1978, 43, 2480.
~ l l i a m s , D. R.; Harigaya, Y.; Moore, J. L.; D'sa, A J. Am. Chem. Soc., 1984, 706, 2641.
a) Fleming, 1. in Comprehensive Organic Chemistry. B.M. Trost, 1. Fleming Éd, Pergamon Press, Oxford, l991, Vol. 2, 563; b) Coni, G.; Tagliavini, E.; Urnani-Ronchi, A Gazz. Chim. Ital., 1997, 727, 247. Hosomi, A; Sakurai, H. Tetrahedron Left., 1976, 1295. a) Danishefsky, S. J.; DeNinno, M. P.; Phillips, G. B.; Zelle, R. E.; Lartey, P. A. Tetrahedron, 1986, 42, 2809; b) Danishefsky, S.; DeNinno, M.
Tetrahedron Lett, 1985, 26, 823. Hooz, J.; Gilani, S. S. Can. J. Chem., 1968, 46, 86. Downie, L. M.; Holmes, J. B.; Lee, J. B. Chern&/nd., 1966, 900. a) Diver, S.T. in Encyclopedia of Reagenfs for Organic Synthesis, L. A Paquette Éd, Wiley 8 Sons, Chichester, 1995, Vol. 7, 5014; b) Cobb, J.
E. in Encyclopedia of Reagenfs for Organic Synthesis, L. A Paquette Éd,
Wiley 8 Sons, Chichester, 1995, Vol. 8, 5357. Tsushima, K.; Murai, A Tetrahedron Lett., 1992, 33, 4345.
Boukouvalas, J.; Fortier, G., Radu, 1.-1. J. Org. Chem., 1998, 63, 916. Lee, E.; Yoo, S.-K.; Choo, H.; Song, H. Y. Tetrahedron Lett, 1998, 39,
31 7. Suzuki, T.; Koisumi, K.; Suzuki, M.; Kurosawa, E. Chem. Left., 1983, 1639.
Barrow, R. A; Capon, R. J. Aust. J. Chem., 1990, 43, 895. Capon, R. J.; Barrow, R. A; Rochfort, S.; Jobling, M.; Skene, C.; Lacey, E.; Gill, J. H.; Friedel, T.; Wadsworth, D. Tetrahedron, 1998, 54, 2227. Au moment de terminer la version finale de ce document, une autre synthèse a été publiée: Wang, Y.-M.; Shen, M. J. Org. Chem., 1998, 63,
1414. Hatakeyama, S.; Sakurai, K.; Saijo, K.; Takano, S. Tetrahedron Lett., 1985, 26, 1333.

Gurjar, M. K; Mainkar, P. S. Heterocycles, 1990, 37, 407.
Chikashita, H.; Nakamura, Y.; Uemura, H.; Itoh, K. Chem. Leff., 1993. 477. Capon, R. J.; Barrow, R. A J. Org. Chem., 1998, 63,75. Capon, R. J.; Barrow, R. A; Skene, C.; Rochfort, S. Tetrahedron Lett , 11997, 38, 7609. a) Koert, U.; Stein, M.; Wagner, H. Liebigs Ann. Chem., 1995, 141 5; b) Jefford, C. W.; Jaggi, D.; Boukouvalas, J. Tetrahedron Lett, 1986, 27, 401 1.
a) Takaki, K. S. in Encyclopedia of Reagents for Organic Synthesis, L. A Paquette Éd, Wiley 8 Sons, Chichester, 1995, Vol. 6, 4185; b) Brown, C. A; Jadhav, P. K. Org. Synth., 1987, 65, 224; c) Brown, C. A; Yamashita,
A J. Chem. Soc. Chem. Commun., 1976, 959; d) Brown, C. A. ; Yarnashita, A; J. Am. Chem. Soc., 1975. 97, 891.
Tellier, F.; Harnmound, A; Ratovelomanana, V.; Linstrumelle, G.; Descoins, C. Bull. Soc. Chim. Fr., 1993, 130, 281. Jacobsen, E. N.; Deng, L. J. Org. Chem., 1992, 57, 4320.
Jacobson, M.; Keiser, 1.; Chambers, D. L.; Miyashita, D. H.; Harding, C. J.
Med. Chem., 1971, 14, 236. Chen, S.-Y.; Jouillé, M. M. Synth. Commun., 1984, 14, 591. Enders, D.; Finkam, M. Liebigs Ann. Chem., 1993, 551. Varoglu, M.; Peters, B. M.; Crews, P. J. Nat. Prod., 1995, 58, 27. Patil, A O.; Freyer, A J.; Carte, B.; Johnson, R. K.; Lahouratate, P. J. Nat. Prod., f 996, 59, 219.
Tomioka, K.; Cho, Y.-S.; Sato, F.; Koga, K. J. Org. Chem., 1988, 53,
4094, a) Caine, D. in Comprehensive Organic Chemistry, B. M. Trost, 1. Fleming Éd, Pergamon Press, Oxford, 1991, Vol. 3, 1; b) Dykstra, R. R. in
Encyclopedia of Reagents for Organic Synfhesis, L. A Paquette Éd, Wley 8 Sons, Chichester, 1995, Vol. 4, 2668; c) Galiano-Roth, A. S.; Collum, D. B. J. Am. Chem. Soc., 1989, 711, 6772; d) Romesberg, F. E.; Gilchrist, J. H.; Harrison, A T.; Fuller, D. J.; Collum, D. B. J. Am. Chem. Soc., 1991, 113, 5751. Dess, D. B.; Martin, J. C. J. Org. Chem., 1983, 48, 41 55.
a) Ireland, R. E.; Liu, L. J. Org. Chem., 1993, 58, 2899; b) Dess, D. B.; Martin, J. C. J. Am. Chem. Soc., 1991, 113, 7277.

Imamoto, T.; Takiyama, N.; Nakamura, K. Tetrahedmn Lett, 1985, 26, 4763.
Bartoli, G.; Bosco, M.; Beek, J. V.; Sambri, L.; Marcantoni, E. Tetrahedmn Lett., 1996, 37, 2293. Verboom, W.; Visser. G. W.; Reinhoudt, D. N-Synthesis, 1981, 807.
Katsuki, T.; Sharpless, K B. J. Am. Chem. Soc., 1980, 102, 5974. a) Johnson, R. A; Sharpless, K B. in Comprehensive Organic Chemistry. B. M. Trost, i. Fleming Éd, Pergamon Press, Oxford, 1991. Vol. 7, 389; b)
Rossiter, B. E. in Asymmetnc Synthesis, J. D. Momson, J. W. Scott Éd, Acadernic Press, Orlando, 1 984, Vol. 5, 1 93. Chin, C. S.; Lee, B.; Kim, S.; Chun, J. J. Chem. Soc. Dalton Trans., 1991, 443.
a) Magnus, P.; Mendoza, J. S.; Stamford, A; Ladlow, M.; Willis, P. J. Am. Chem. Soc., 1992, 7 74, 10232; b) Kuehne, M. E.; Matson, P. A.;
Bornmann, W. G. J. Org. Chem., 1991, 56, 51 3.
a) Ward, D. E.; Rhee, C. K. Tetrahedron Lett., 1991, 32, 7165; b) Dale, J. A; Mosher, H. S. J. Am. Chem. Soc., 1973, 95, 512; c) Dale, J. A; Dull, D. L.; Mosher, H. S.; J. Org. Chem., 1969, 34,2543. Hill, J. G.; Rossiter, B. E.; Sharpless, K. B. J. Org. Chem., 1983, 48, 3607.
Strekowski, L.; Visnick, M.; Battiste, M. A J. Org. Chem., 1986, 57, 4836.
a) Davis, F. A; Haque, M. S. J. Org. Chem., 1986, 57, 4083, b) Davis, F. A; Haque, M. S.; Utlatowski, T. G.; Towson, J. C. J. Org. Chem., 1986, 51, 2402.
a) Chen, B.-C.; Murphy, C. K.; Kumar, A; Reddy, R. T.; Clark, C.; Zhou, P.; Lewis, B. M.; Gala, O.; Mergelsberg, 1.; Scherer, D.; Buckley, J.; DiBenedetto, D.; Davis, F. A Org. Synth., 1996, 73, 159; b) Bulman Page, P. C.; Heer, J. P.; Bethell, D.; Lund, A; Collington, E. W.; Andrews, D. M. J. Org. Chem., 1997, 62, 6093.
Davis, F. A; Reddy, G. V.; Chen, B.-C.; Kumar, A; Haque, M. S. J. Org. Chem., 1995, 60, 6148. Suter, P., mémoire de maitrise à venir, Université Laval, 1998.
Eliel, E. L. ; Wilen, S. H. in Stereochemistry of Organic Compounds, Wiley&sons, New York, 1994, 381. Hickman, J. R.; Kenyon, J. J. Chem. Soc., 1955, 2051. a) Hanessian, S. Total Synthesis of Natural Products: The 'Chiron' Approach, Pergamon Press, Oxford, 1983; b) Scott, J. W. in Asymmefnc

Synthesis, J. D. Momson, J. W. Scott Éd, Academic Press, Orlando, 1984, vo1.4, 1. Chen, S.-T.; Fang, J.-M. J. Org. Chem., 1997, 62, 4349. Pour des exemples récents voir: a) Dosa, P. 1.; Fu, G. C. J. Am. Chem. Soc., 1998, 120, 445; b) Bartoli, G.; Bosco, M.; Sambri, L.; Marcantoni, E. Tetrahedron LeK, 1997, 38, 3785. Kolb, H. C.; Van Nieuwenhze, M. S.; Sharpless, K. B. Chem. Rev., 1994,
94, 2483. Mukaiyama, T.; Yamada, T.; Hagata, T.; Imagawa, K. Chem. Left., 1993, 327. Guo, J.; Du@, K- J.; Stevens, K. L.; Dalko, P. 1.; Roth, R. M.; Hayward, M. M.; Kishi, Y. Angew. Chem. /nt. Ed. Engl., 1998, 37, 187. Walker II, J. A; Chen, J. J.; Wise, 0. S.; Towsend, L. B. J. Org. Chem., 1996, 61, 2219. Sharpless, K. B.; Amberg, W.; Bennani, Y. 1.; Crispino, G. A; Hartung, J.;
Jeong, K.S.; Kwong, H.-L.; Morikawa, K.; Wang, L M . ; Xu, D.; Zhang, X.- L. J. Org. Chem., 1992, 57, 2768. Curran, D. P.; Ko, S.-B. J. Org. Chem., 1994, 59, 6139.
Boukouvalas, J.; Cheng, Y.-X.; Fortier, G.; Suter, P., tapuscrit en préparation. Still, W. C.; Kahn, M.; Mitra, A J. Org. Chem., 1978, 43, 2923. Perrin, D. D.; Armarego, W. L. F. Purification of Laboratory Chemicals, 3e Éd., Pergamon Press, Oxford, 1988.
IMAGE EVALUATION
APPLIED 1 IMAGE. lnc - - - 1653 East Main Street - -- - Rochester. NY 14609 USA -- - Phone: 71 W82-0300 -- -- - - Fax: 71 6/288-5989
O 1993. Applied Image. lm.. AU Fùghts Resewed





























