Embed Size (px)
Citation preview

Hana ManceauMCU PH
Service de Biochimie Clinique, Hôpital Beaujon Inserm U1149 - Université Paris Diderot
Faculté Médecine - Université Paris Diderot
L2-UE8 – 2018/19

Connaissances PACES : formules des AA propriétés physico-chimiques des AA, protéines protéosynthèse
Objectifs L2 : différentes composantes du métabolisme protéique
et ses finalités éléments de régulation métabolique moyens d'exploration de ce métabolisme déficits enzymatiques du cycle de l’urée
Prérequis et objectifs
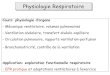
Protéines - macromolécules comportant de l'azote et composées d'une séquence d'acides aminés reliés par des liaisons peptidiques
Rappels
AA essentiels(indispensables)
AA semi-indispensables (chez enfant et allaitement)
LeucineThréonine
LysineTryptophane
Phénylalanine Valine
MéthionineIsoleucine
GlutamineTyrosineCystéineArginineHistidine
C CN
H
R1
O
H
H C CN
H
R2 O
OH
H
0%
20%
40%
60%
80%
100%
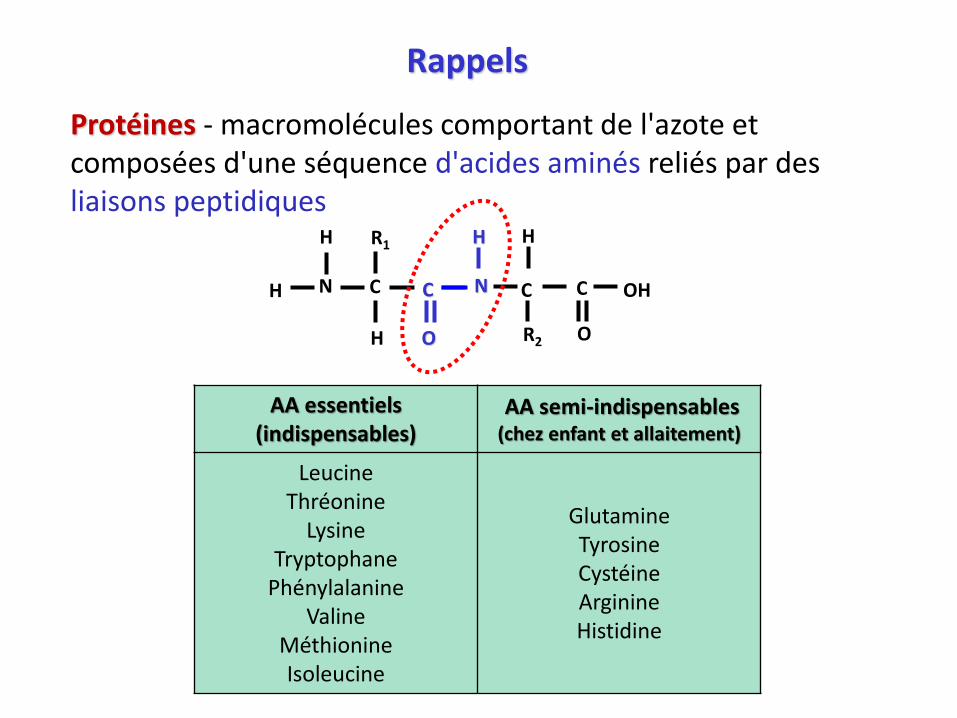
Eau extracellulaire (25% 20 L)
Eau intracellulaire (37% 25 L)
Minéraux (6% 4 kg)
Graisse (10-30% 10 kg)
Masse corporelle 70 kg
Masse maigre
Masse grasse
Protéines (16% 10 kg)
Protéines - importance vitale
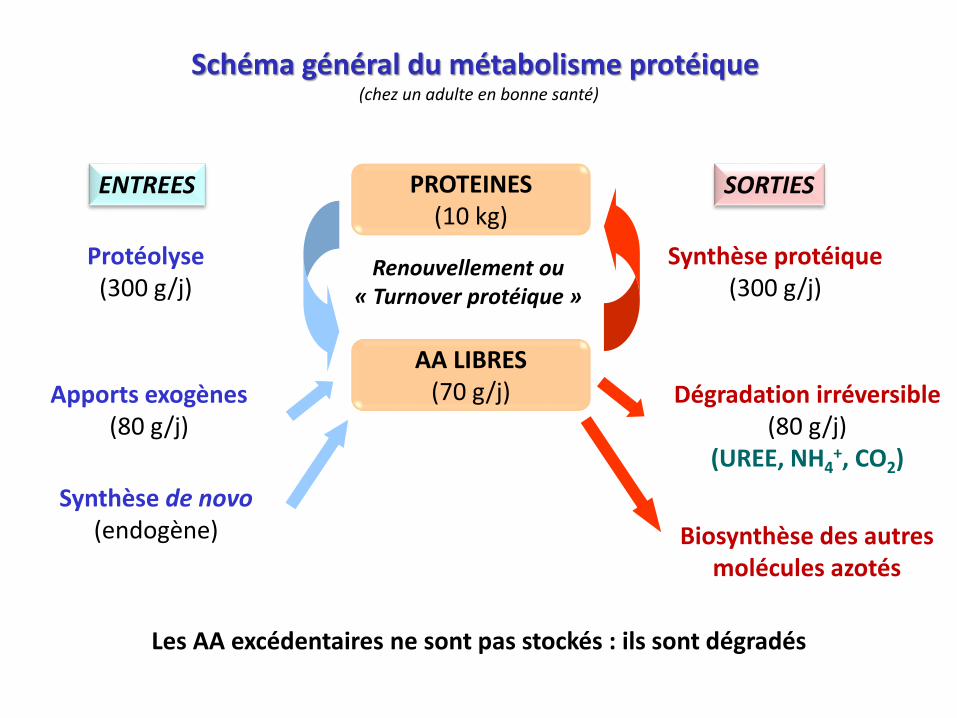
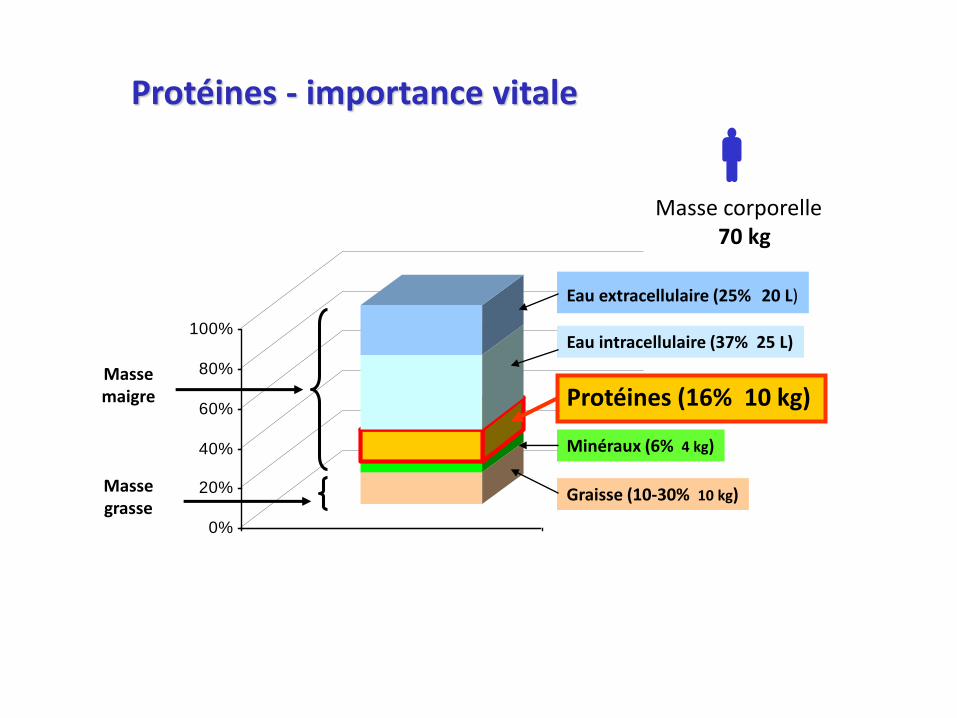
Schéma général du métabolisme protéique (chez un adulte en bonne santé)
PROTEINES(10 kg)
Synthèse protéique(300 g/j)
Biosynthèse des autres molécules azotés
Apports exogènes(80 g/j)
Synthèse de novo(endogène)
Dégradation irréversible(80 g/j)
(UREE, NH4+, CO2)
Renouvellement ou« Turnover protéique »
Les AA excédentaires ne sont pas stockés : ils sont dégradés
AA LIBRES(70 g/j)
Protéolyse(300 g/j)
ENTREES SORTIES
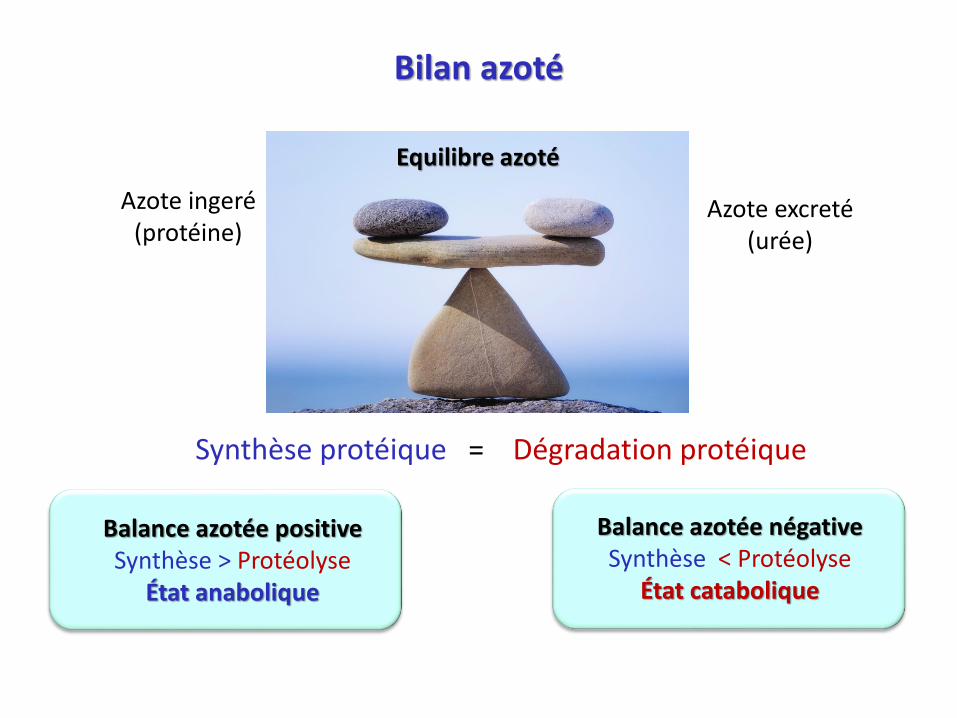
Bilan azoté
Balance azotée positiveSynthèse > Protéolyse
État anabolique
Azote ingeré(protéine)
Azote excreté(urée)
Equilibre azoté
Synthèse protéique = Dégradation protéique
Balance azotée négativeSynthèse < Protéolyse
État catabolique
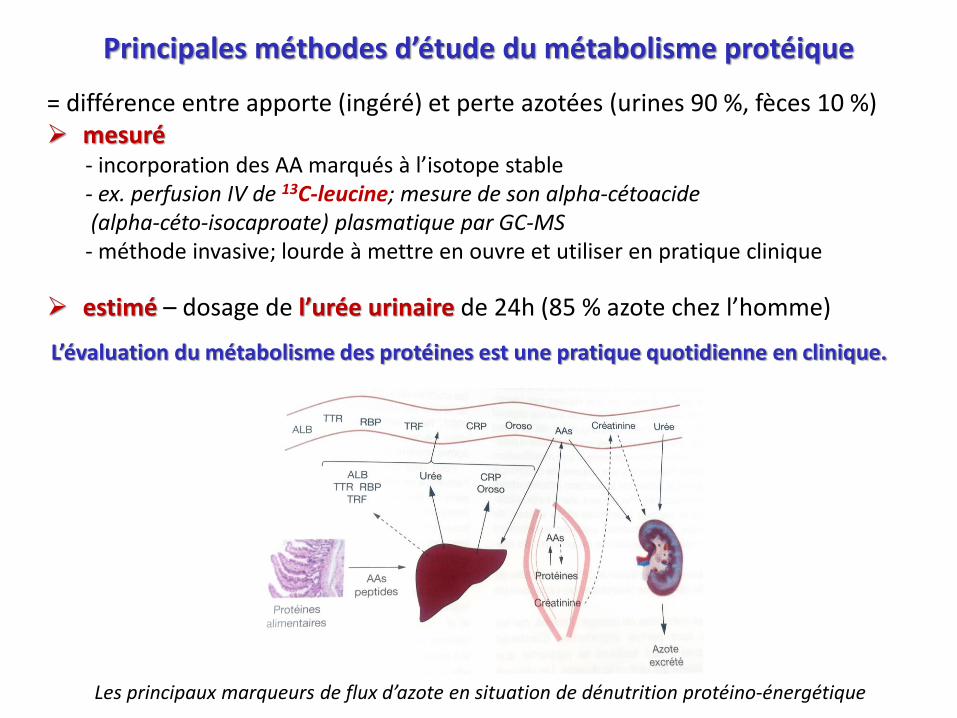
Principales méthodes d’étude du métabolisme protéique
= différence entre apporte (ingéré) et perte azotées (urines 90 %, fèces 10 %) mesuré
- incorporation des AA marqués à l’isotope stable- ex. perfusion IV de 13C-leucine; mesure de son alpha-cétoacide(alpha-céto-isocaproate) plasmatique par GC-MS
- méthode invasive; lourde à mettre en ouvre et utiliser en pratique clinique
estimé – dosage de l’urée urinaire de 24h (85 % azote chez l’homme)
L’évaluation du métabolisme des protéines est une pratique quotidienne en clinique.
Les principaux marqueurs de flux d’azote en situation de dénutrition protéino-énergétique

Renouvellement des protéines
Protéines participent de façon très variable au renouvellement protéique global en fonction de :
importance quantitative de la protéine (muscles, foie, intestin, peau)
rapidité de renouvellement de protéine
10% 20 %
Variations du renouvellement protéique
Age - NNé 15 g/kg/j >> adulte 4 g/kg/j synthèse >> protéolyse
Etat nutritionnel - au cours du jeûne protéolyse > synthèse
Etat pathologique (syndrome inflammatoire, traumatisme, sepsis, brûlés)
- situations cataboliques
- renouvellement 3-4x mais pas de gain protéique!
- foie+++ - synthèse des protéines de l’inflammation
- muscle produit des AA protéolyse > synthèse

Variations physiologiques et pathologiques des ENTREES
Alimentation = Apport des AA exogènes
Correspond à l’apport alimentaire en protéines qui subissent leur digestion
au niveau du tractus digestif
I. Aspects quantitatifs
Chez l’adulte, en pays développé, apports 70-100 g/j
Besoins recommandés:
55 g 45 g 110 g
4-6 mois
Dépendent de - l’apport par les autres nutriments- l’activité physique- l’âge et du sexe

Kwashiorkor
Enfants de 6 mois-3ansau moment du sevrage
Pays en voie de développementAnorexie
Opérés, cancéreux, brûlésPerte protéique >> besoins
Carence protéique
Marasme nutritionnel
Carence globale(P, vitamines, minéraux)
En pathologie : MALNUTRITION PROTEINO-CALORIQUE

PROTEOLYSE = catabolisme protéique
Source principale d’AA pour l’organisme (75 %)
Multiples fonctions :
« Ménage cellulaire » (renouvellement basal des protéines,
élimination des protéines anormales)
Genèse des peptides antigéniques (réponse immunitaire)
Production d’énergie en situation de carence (muscle)
Régulation de l’abondance tissulaire des enzymes
Consomme de l’énergie +++
Régulée +++ par conditions nutritionnelles et hormonales

Systèmes protéolytiques
Système lysosomal : foie, reins +++ (< 15% protéolyse)
- ATP dépendant
Système Calpaïne-Calpastatine : - calcium dépendant - cytosolique
- dégradation des protéines du cytosquelette +++
Système protéasome : muscle +++
dans les états cataboliques +++
- ATP dépendant
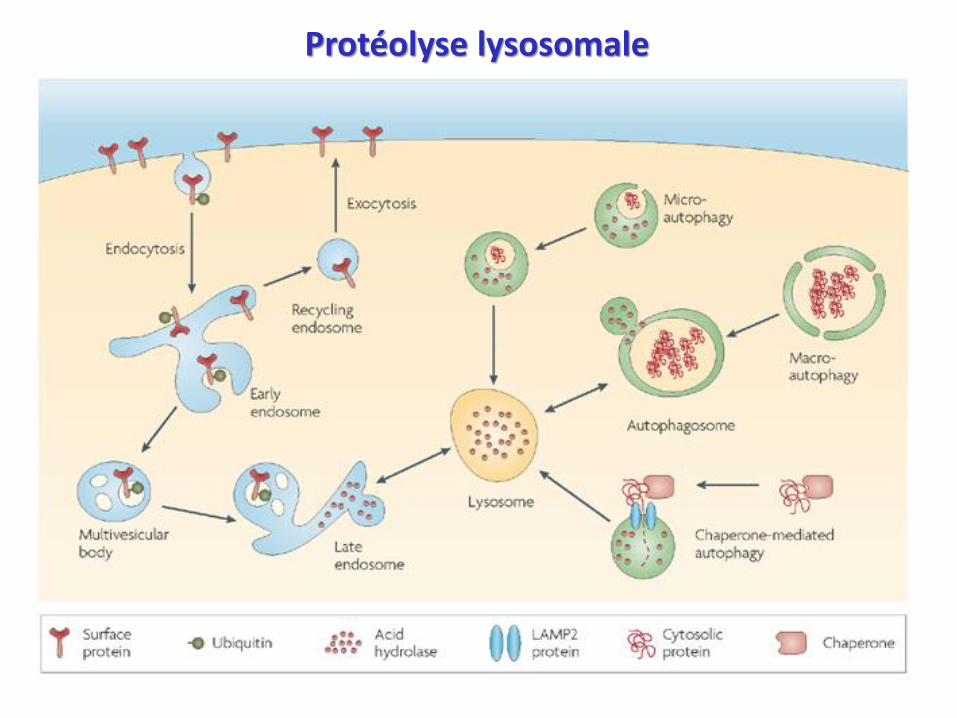
Protéolyse lysosomale

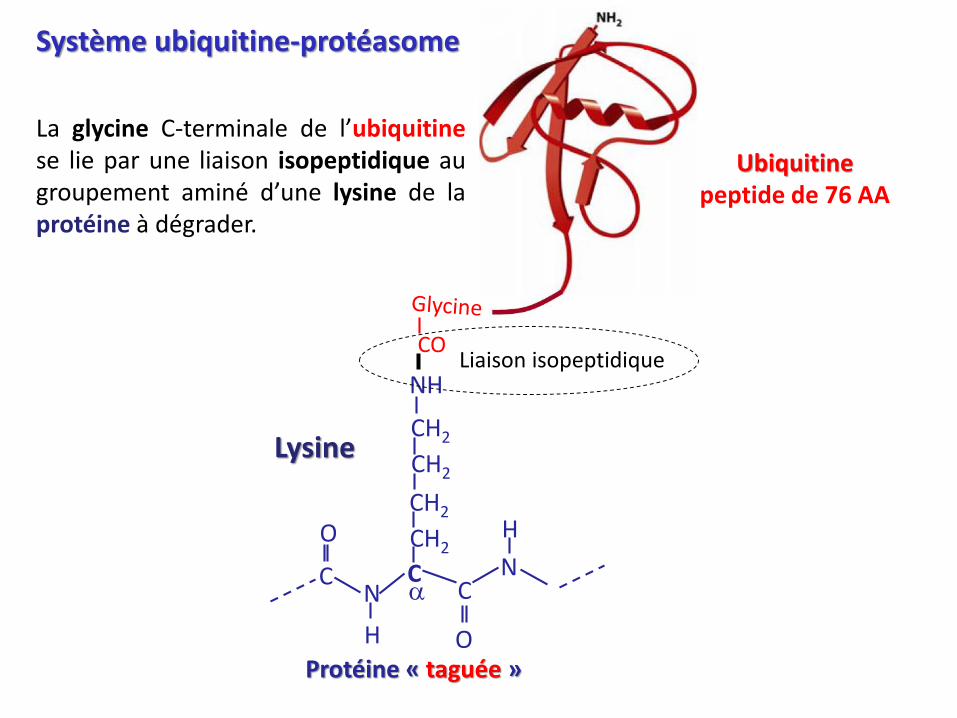
Système ubiquitine-protéasome
• Complexe multienzymatique cytosolique
• Dégradation des protéines intracellulaire
(enzymes, protéines régulatrices)
• Dégradation des protéines anormales
• Majorité de la protéolyse au niveau musculaire +++
• Le protéasome « digère » les protéines marquées par l’ubiquitine
Complexe 20S - activité catalytique protéolytique (CP)
Complexe 19S - activité régulatrice (RP)
O
N
H O
N
H
Protéine « taguée »
CH2
CH2
Lysine
CO
Ubiquitinepeptide de 76 AA
CC
C
CH2
CH2
NHLiaison isopeptidique
a
La glycine C-terminale de l’ubiquitinese lie par une liaison isopeptidique augroupement aminé d’une lysine de laprotéine à dégrader.
Système ubiquitine-protéasome
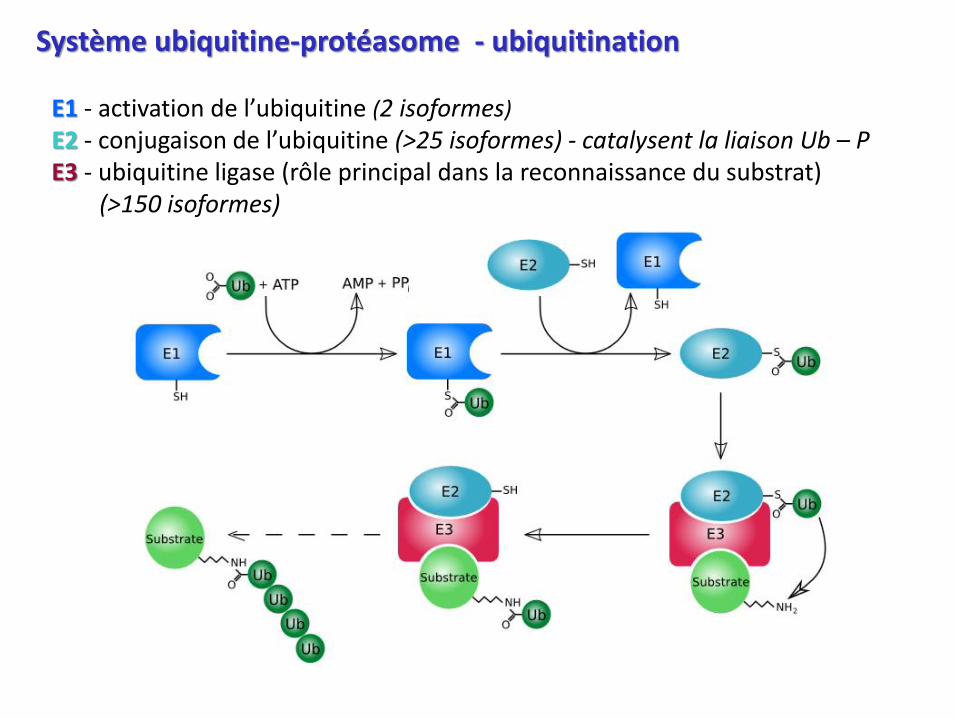
Système ubiquitine-protéasome - ubiquitination
E1 - activation de l’ubiquitine (2 isoformes)
E2 - conjugaison de l’ubiquitine (>25 isoformes) - catalysent la liaison Ub – PE3 - ubiquitine ligase (rôle principal dans la reconnaissance du substrat)
(>150 isoformes)
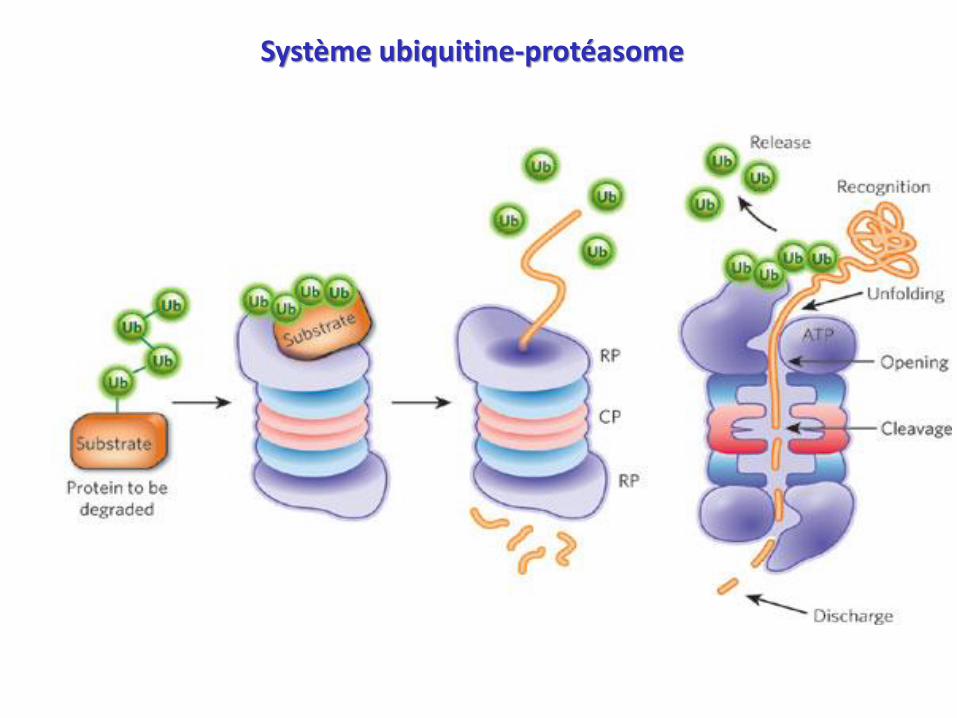
Système ubiquitine-protéasome

AA en position N-terminale :
- stabilisants : Met, Ser, Gly (protéines à demi-vie longue)
- déstabilisants : Arg, Lys, His (protéines à demi-vie courte)
Séquence signal
- courtes séquence spécifique des AA deviendraient exposés au fur et
à mesure du vieillissement de la protéine
- « motifs de destruction » PEST (Pro-Glu-Ser-Thr)
Signaux de la protéolyse
Ciblage en fonction de :- poids moléculaire- degré de glycosylation- point isoélectrique…

Schéma général du métabolisme protéique (chez un adulte en bonne santé)
PROTEINES(10 kg)
Synthèse protéique(300 g/j)
Biosynthèse des autres produits azotés
Apports exogènes(80 g/j)
Synthèse de novo(endogène)
Dégradation irréversible(80 g/j)
(UREE, NH4+, CO2)
Renouvellement ou« Turnover protéique »
AA LIBRES(70 g/j)
Protéolyse(300 g/j)
ENTREES SORTIES
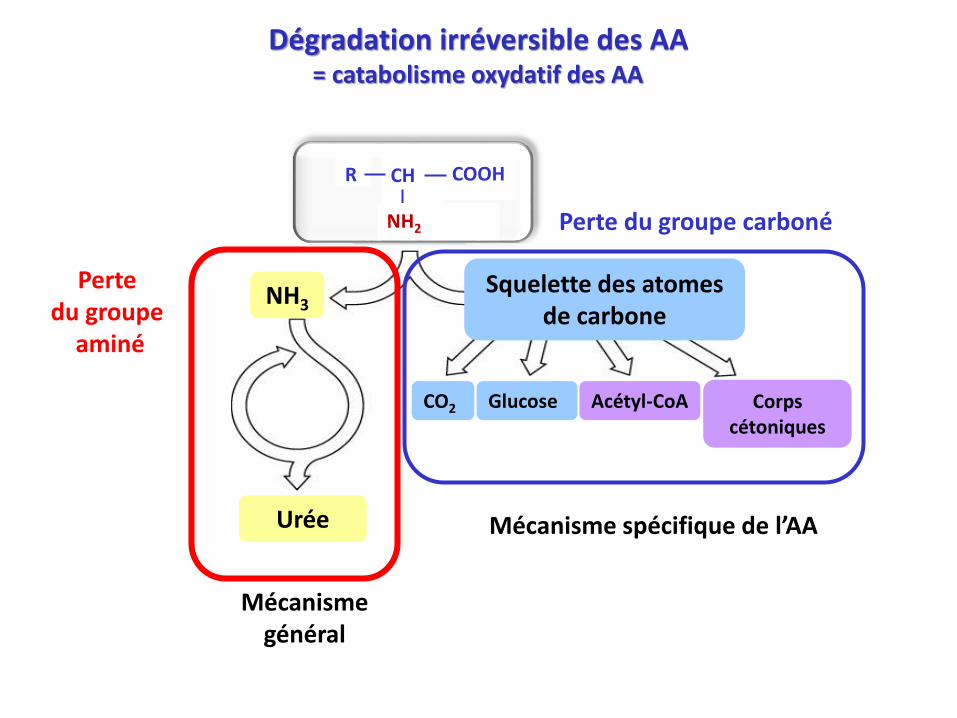
Dégradation irréversible des AA = catabolisme oxydatif des AA
Squelette des atomes de carbone
Glucose Acétyl-CoA Corps cétoniques
Urée Mécanisme spécifique de l’AA
Perte du groupe carboné
Perte du groupe
aminé
Mécanismegénéral
CO2
NH3
COOHCHR
NH2

Urée
Perte du groupe
aminé
Mécanismegénéral
NH3
COOHCHR
NH2
Élimination du groupe aminé des AA
Transaminations
Désaminations oxydatives
Désaminations NON oxydatives
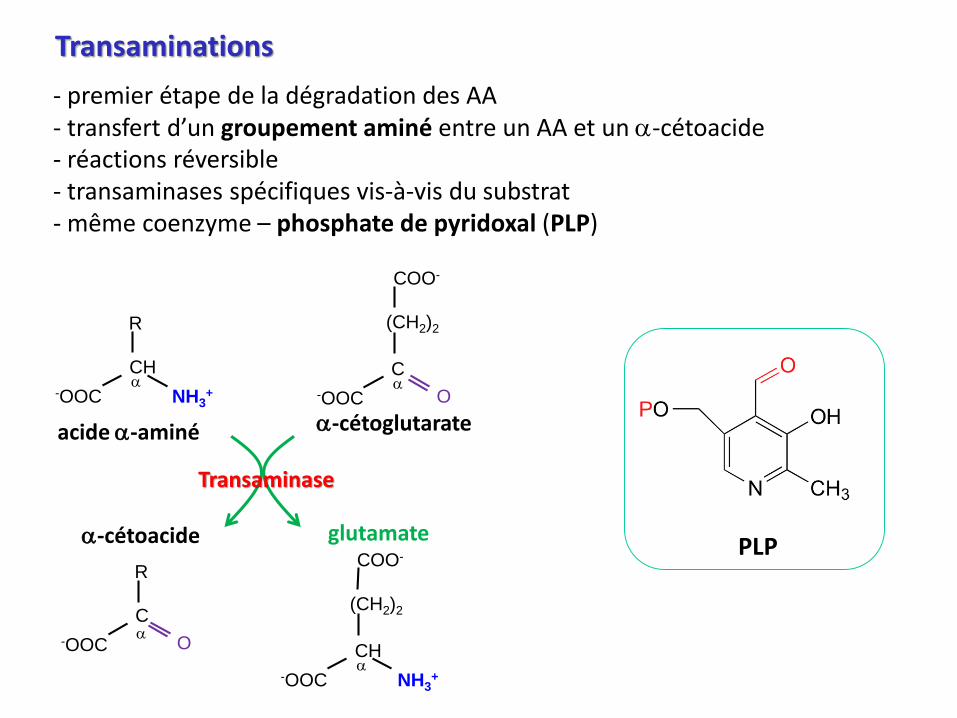
Transaminations
- premier étape de la dégradation des AA- transfert d’un groupement aminé entre un AA et un a-cétoacide- réactions réversible- transaminases spécifiques vis-à-vis du substrat- même coenzyme – phosphate de pyridoxal (PLP)
acide a-aminé
a-cétoacide
a-cétoglutarate
glutamate
Transaminase
R
CH
-OOC NH3+
R
C
-OOC
a
(CH2)2
CH
-OOC NH3+
a
COO-
(CH2)2
C
-OOCa
COO-
O
Oa
PLP
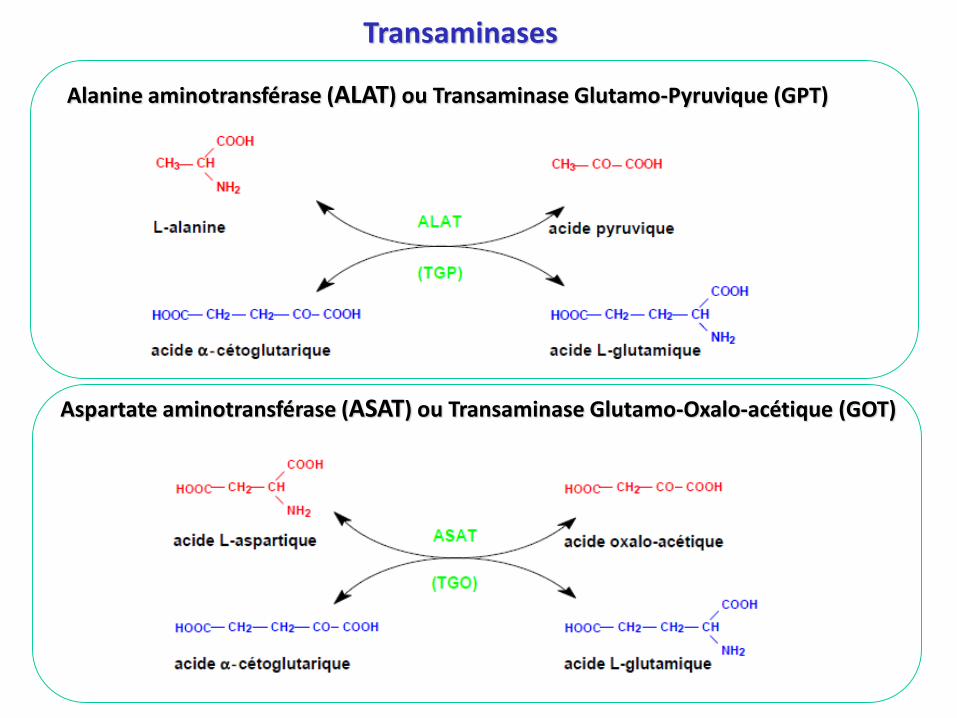
Alanine aminotransférase (ALAT) ou Transaminase Glutamo-Pyruvique (GPT)
Aspartate aminotransférase (ASAT) ou Transaminase Glutamo-Oxalo-acétique (GOT)
Transaminases
Acide a-aminé
a-cétoacide
a-cétoglutarate
glutamate
Transaminase
NAD+ + H2O
Glutamate déshydrogénase
NADH,H+ + NH4+
R
CH
-OOC NH3+
R
C
-OOC
a
(CH2)2
CH
-OOC NH3+
a
COO-
(CH2)2
C
-OOCa
COO-
O
Oa
Mitochondrie (foie)
urée
NH2
C
ONH2
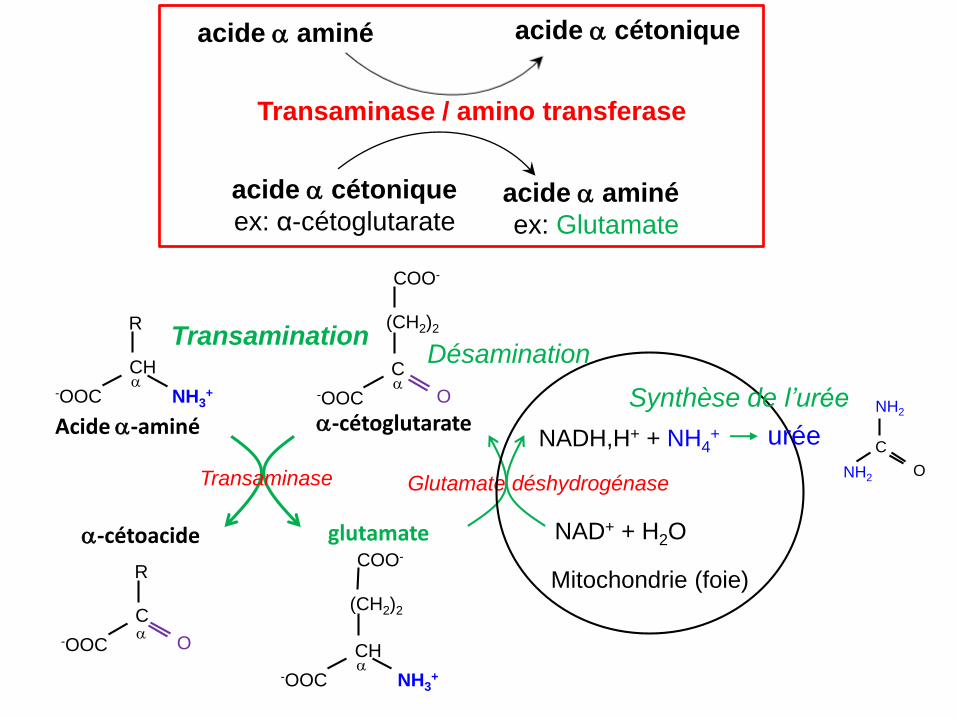
Transamination Désamination
Synthèse de l’urée
acide a cétonique
ex: α-cétoglutarateacide a aminé
ex: Glutamate
Transaminase / amino transferase
acide a aminé acide a cétonique
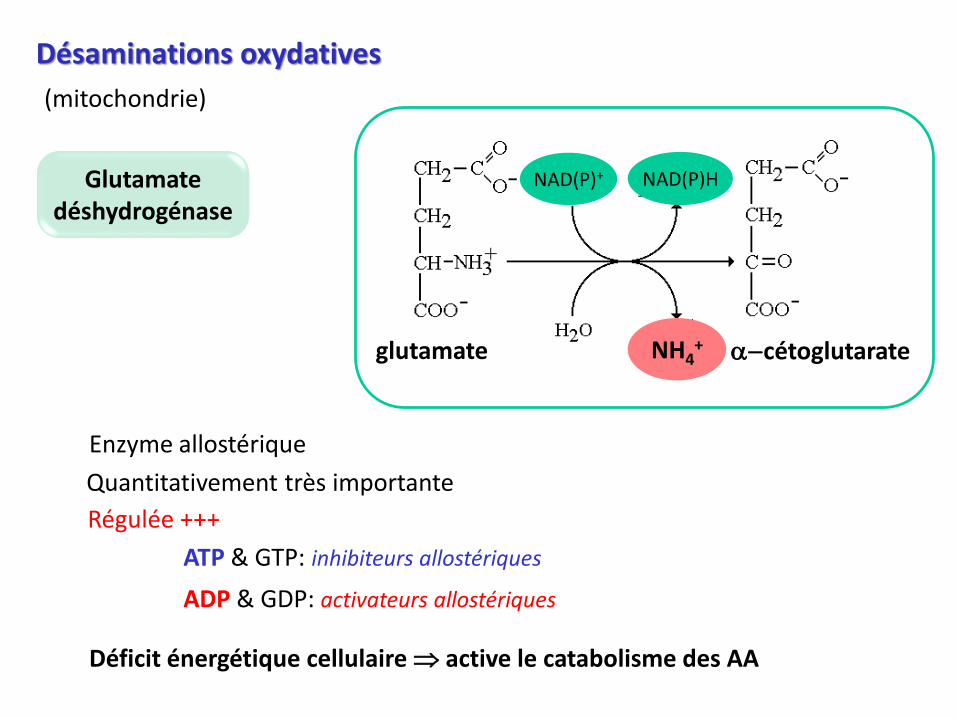
Désaminations oxydatives
glutamate a-cétoglutarateNH4+
NAD(P)+ NAD(P)HGlutamatedéshydrogénase
(mitochondrie)
Quantitativement très importante
Enzyme allostérique
Régulée +++
ATP & GTP: inhibiteurs allostériques
ADP & GDP: activateurs allostériques
Déficit énergétique cellulaire active le catabolisme des AA
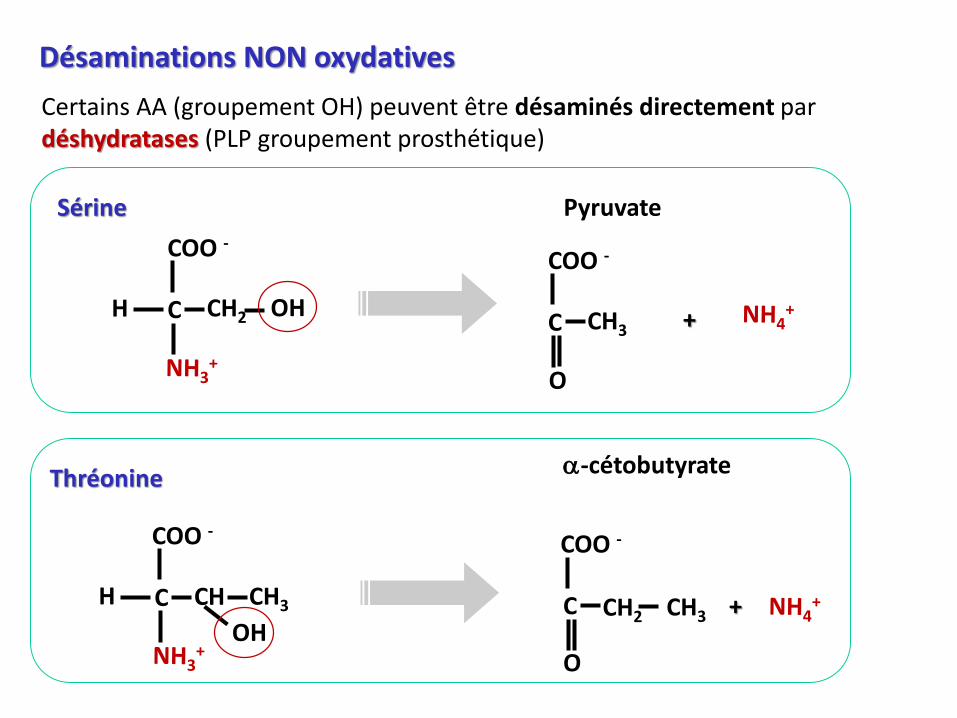
Désaminations NON oxydatives
Certains AA (groupement OH) peuvent être désaminés directement par déshydratases (PLP groupement prosthétique)
PyruvateSérine
COO -
H C
Thréonine
COO -
H C
COO -
C
COO -
C
a-cétobutyrate
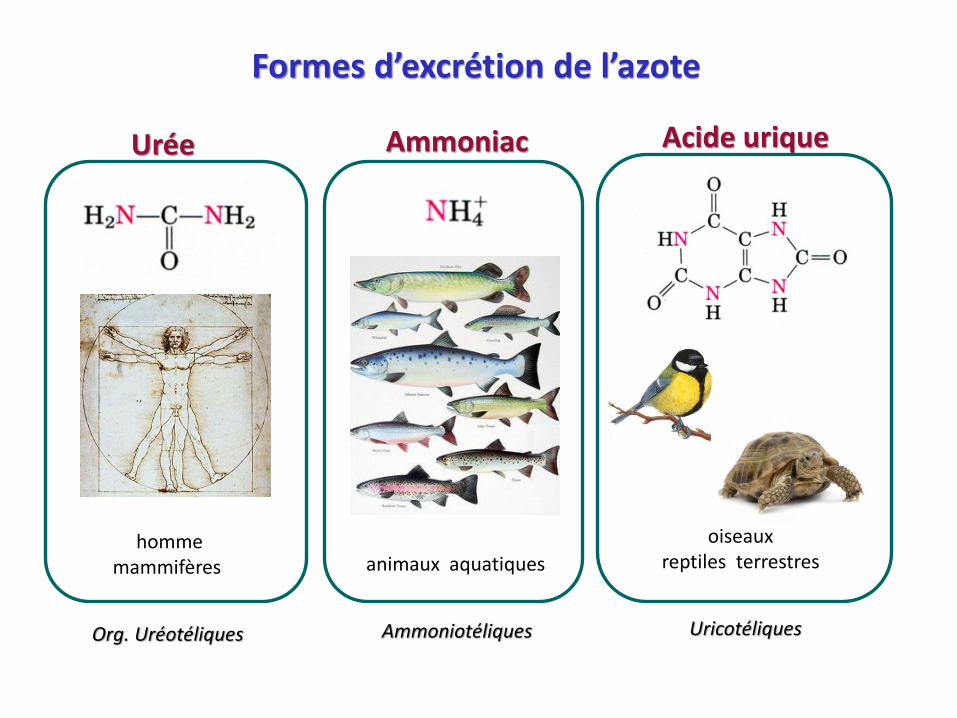
Formes d’excrétion de l’azote
AmmoniacUrée Acide urique
hommemammifères
oiseauxreptiles terrestres animaux aquatiques
AmmoniotéliquesOrg. Uréotéliques Uricotéliques
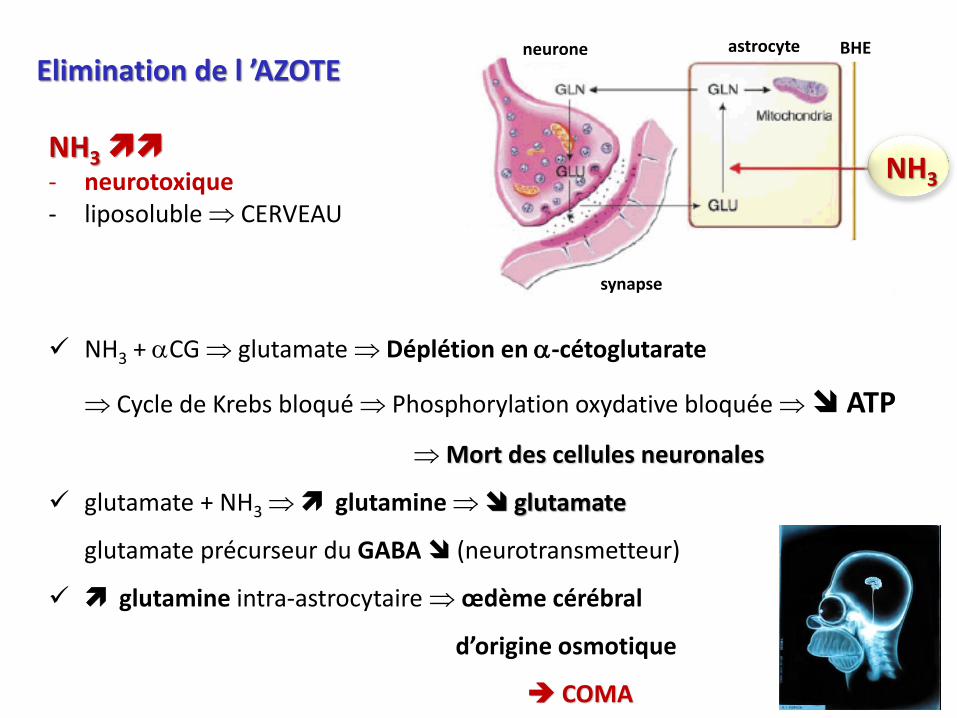
Elimination de l ’AZOTE
NH3
- neurotoxique- liposoluble CERVEAU
NH3 + aCG glutamate Déplétion en a-cétoglutarate
Cycle de Krebs bloqué Phosphorylation oxydative bloquée ATP
Mort des cellules neuronales
glutamate + NH3 glutamine glutamate
glutamate précurseur du GABA (neurotransmetteur)
glutamine intra-astrocytaire œdème cérébral
d’origine osmotique
COMA
NH3
astrocyteneurone BHE
synapse
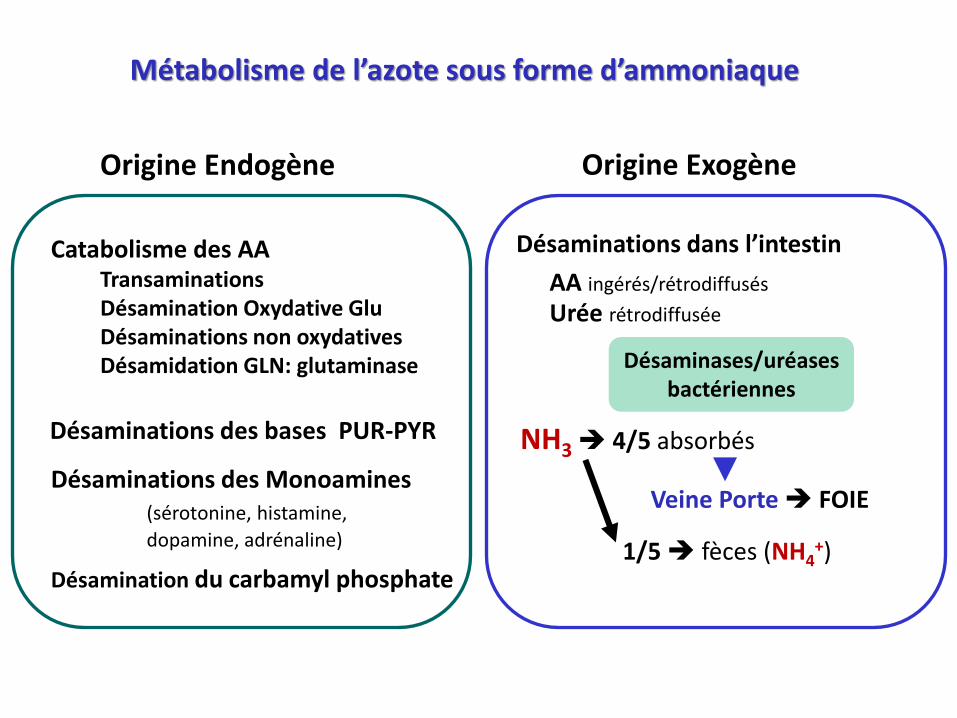
Métabolisme de l’azote sous forme d’ammoniaque
Catabolisme des AATransaminationsDésamination Oxydative GluDésaminations non oxydativesDésamidation GLN: glutaminase
Désaminations des bases PUR-PYR
Désaminations des Monoamines(sérotonine, histamine,
dopamine, adrénaline)
Désamination du carbamyl phosphate
Origine Endogène Origine Exogène
Désaminations dans l’intestin
AA ingérés/rétrodiffusés
Urée rétrodiffusée
Désaminases/uréasesbactériennes
NH3 4/5 absorbés
Veine Porte FOIE
1/5 fèces (NH4+)

2. Dans le Foie: glutamine glutamate + NH4+ UREE +++
Glutaminase
3. Dans les Reins: élimination sous forme NH4+ : NH3 + H+
20% de l’azote urinaire total
1. Tissus périphériques exportent l’azote vers le FOIE
sous forme de glutamine (GLU + NH4+), Glutamine synthétase
sous forme de alanine dans le muscle +++ (Cycle de l’alanine)
Métabolisme de l’azote sous forme d’ammoniaque

AA le plus abondant dans le plasma
Transporteur d’azote entre les tissus
- Foie : Uréogenèse
- Reins : Ammoniogenèse
Utilisée pour la synthèse de nombreux composés +++
- Substrat pour les bases nucléiques
- Précurseur de Pro, Orn, Arg
Glutamine (GLN = Q)
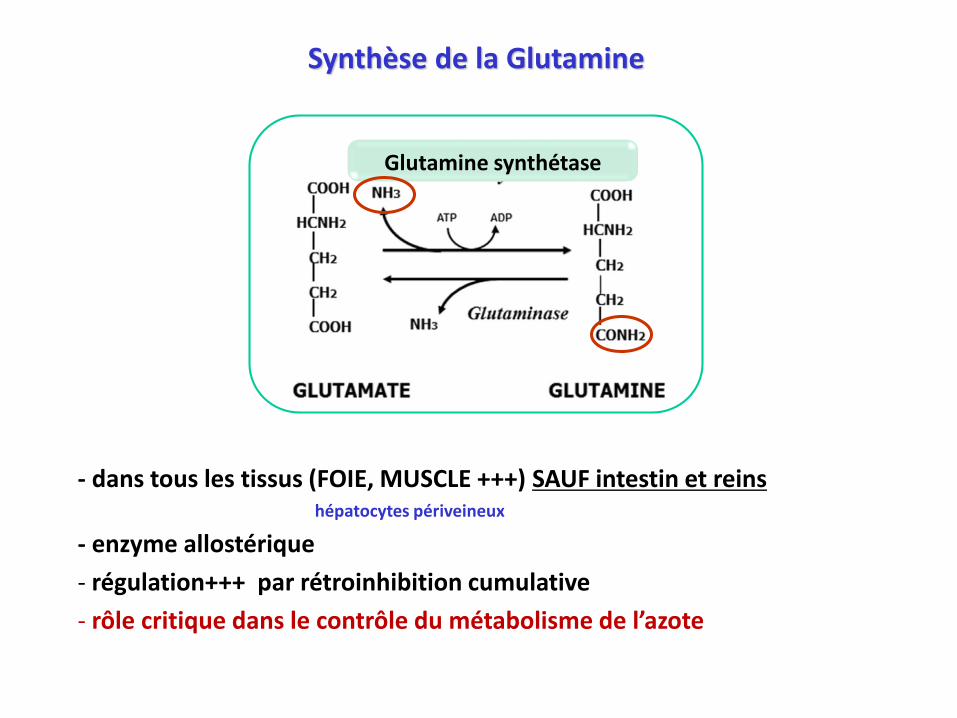
Synthèse de la Glutamine
- dans tous les tissus (FOIE, MUSCLE +++) SAUF intestin et reinshépatocytes périveineux
- enzyme allostérique
- régulation+++ par rétroinhibition cumulative
- rôle critique dans le contrôle du métabolisme de l’azote
Glutamine synthétase
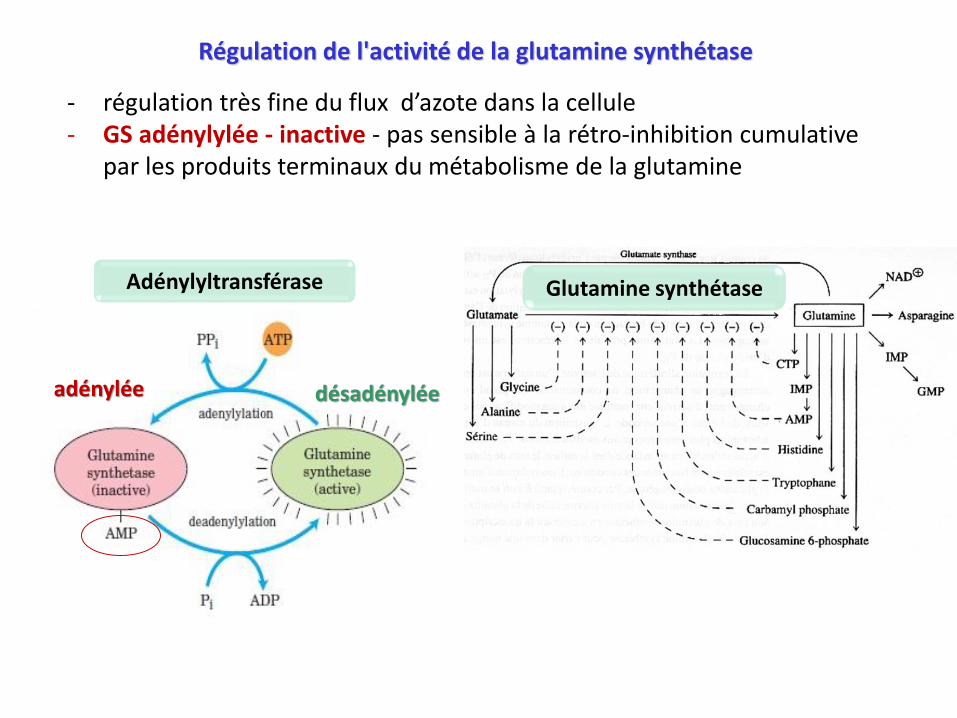
Régulation de l'activité de la glutamine synthétase
- régulation très fine du flux d’azote dans la cellule- GS adénylylée - inactive - pas sensible à la rétro-inhibition cumulative
par les produits terminaux du métabolisme de la glutamine
désadénylée
Adénylyltransférase
adénylée
Glutamine synthétase
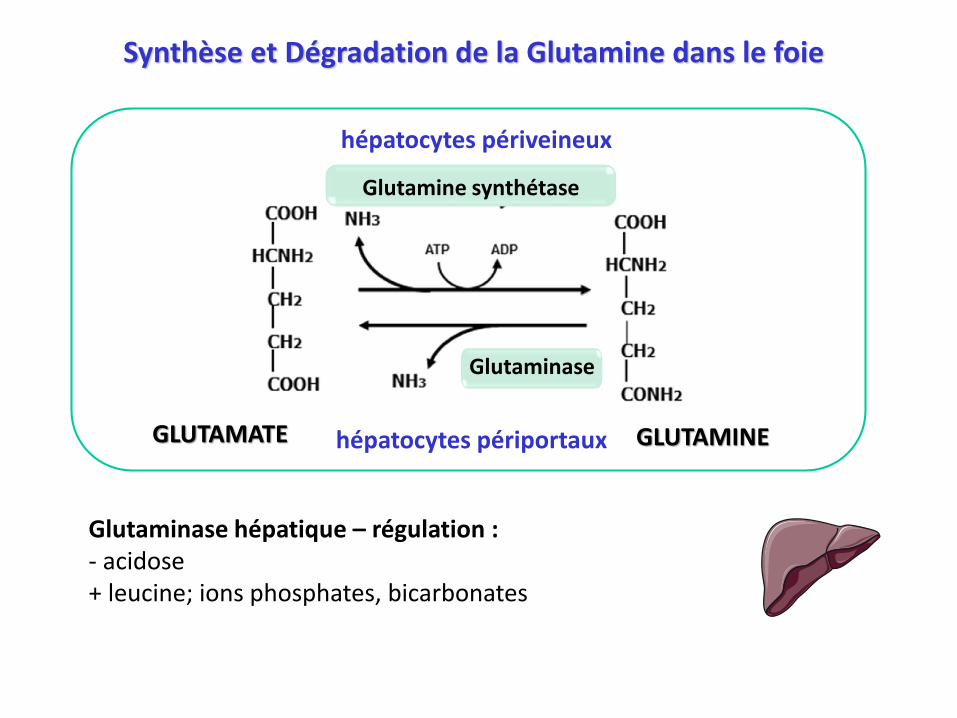
Synthèse et Dégradation de la Glutamine dans le foie
Glutamine synthétase
hépatocytes périveineux
Glutaminase
hépatocytes périportauxGLUTAMATE GLUTAMINE
Glutaminase hépatique – régulation :- acidose+ leucine; ions phosphates, bicarbonates
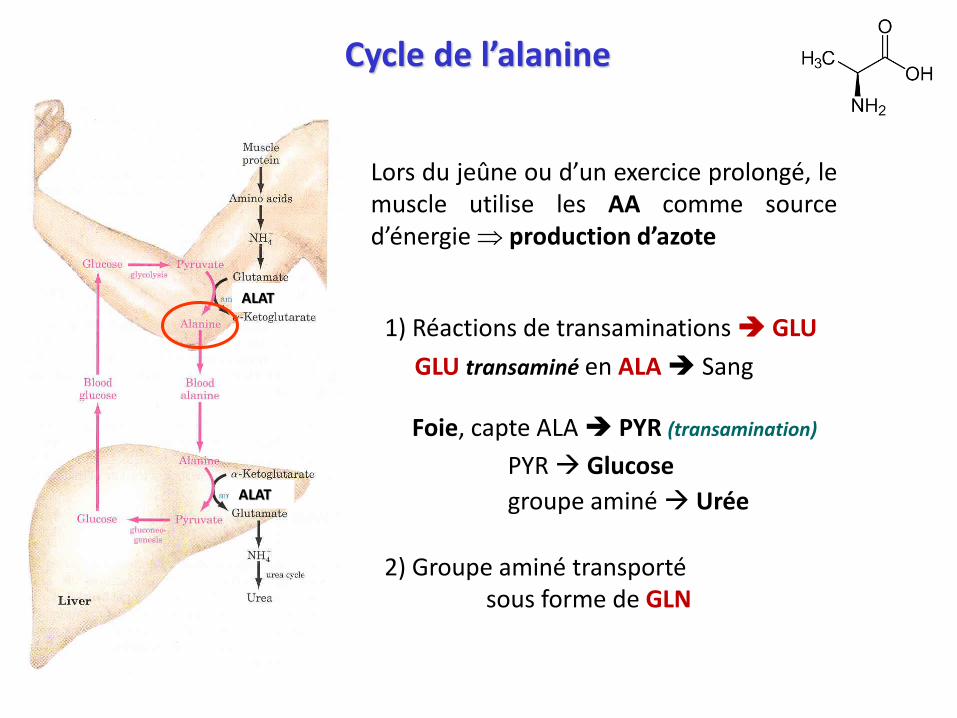
Cycle de l’alanine
Lors du jeûne ou d’un exercice prolongé, lemuscle utilise les AA comme sourced’énergie production d’azote
1) Réactions de transaminations GLU
GLU transaminé en ALA Sang
Foie, capte ALA PYR (transamination)
PYR Glucose
groupe aminé Urée
2) Groupe aminé transportésous forme de GLN
ALAT
ALAT
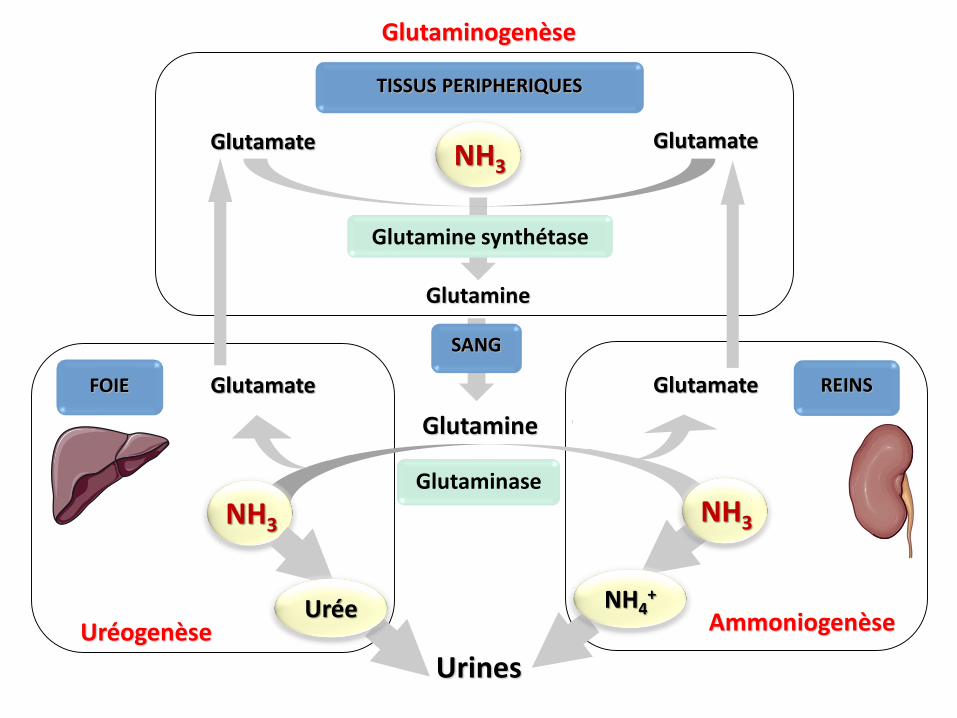
Glutamine
Glutamine
Glutamine synthétase
UrinesUréogenèse Ammoniogenèse
FOIE REINS
Glutaminase
Glutaminogenèse
NH3
Glutamate
Glutamate Glutamate
Glutamate
SANG
TISSUS PERIPHERIQUES
NH3 NH3
NH4+
Urée

cycle Krebs-Henseleit (1932); cycle de l'ornithine; uréogenèse
voie préférentielle d’élimination de l’azote en excès (85%)
élimination de l'ammoniac issu de la dégradation des AA
formation d’une molécule d’urée et régénération de l’ornithine
chez les mammifères ce cycle se déroule uniquement dans le foie
(hépatocytes périportaux)
3 réactions ont lieu dans la matrice mitochondriale et
3 autres se déroulent dans le cytosol
transporteurs entre la matrice mitochondriale et le cytosol :
- 2 transporteur citrulline - ornithine
- 1 translocase glutamate - aspartate
C
O
NH2NH2
Cycle de l’URÉE
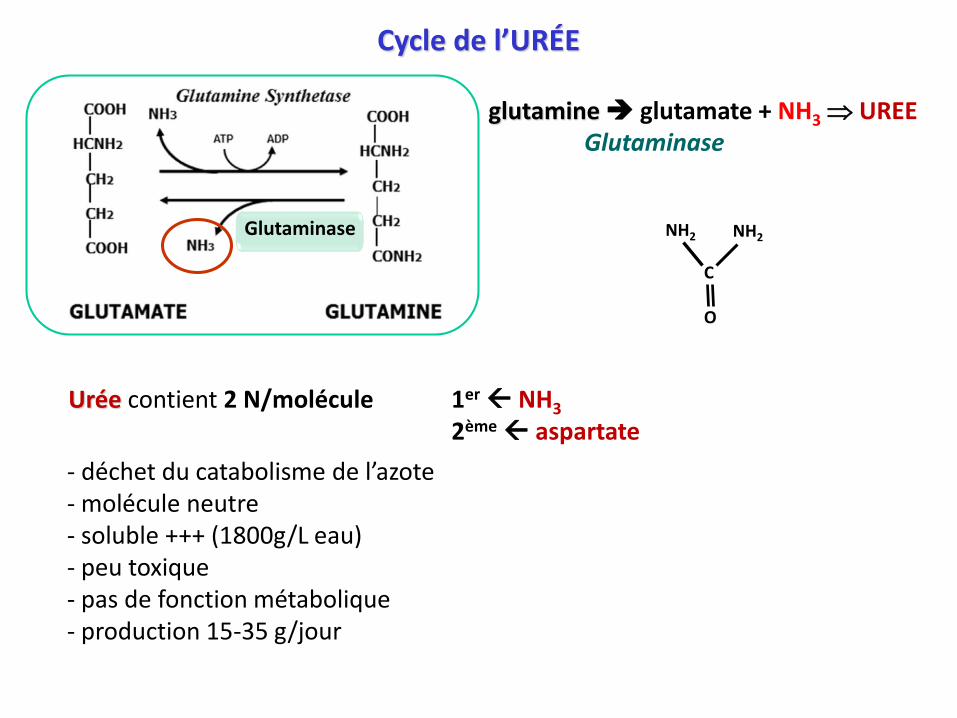
Cycle de l’URÉE
Urée contient 2 N/molécule 1er NH3
2ème aspartate
- déchet du catabolisme de l’azote- molécule neutre - soluble +++ (1800g/L eau)- peu toxique - pas de fonction métabolique- production 15-35 g/jour
C
O
NH2NH2
glutamine glutamate + NH3 UREE Glutaminase
Glutaminase

Ornithine
Citrulline
Carbamylphosphate
NH3HCO3
-
ATP
NAG
Glutamate Acetyl-CoA
NAGS
CPS-I
OTC
mitochondrie
Cycle de l’URÉE – étapes mitochondriales
N Acetyl Glutamate Synthase
Carbamoyl Phosphate Synthétase I
Ornithine Trans Carbamylase
NAGS
CPS-I
OTC
Carbamyl phosphate
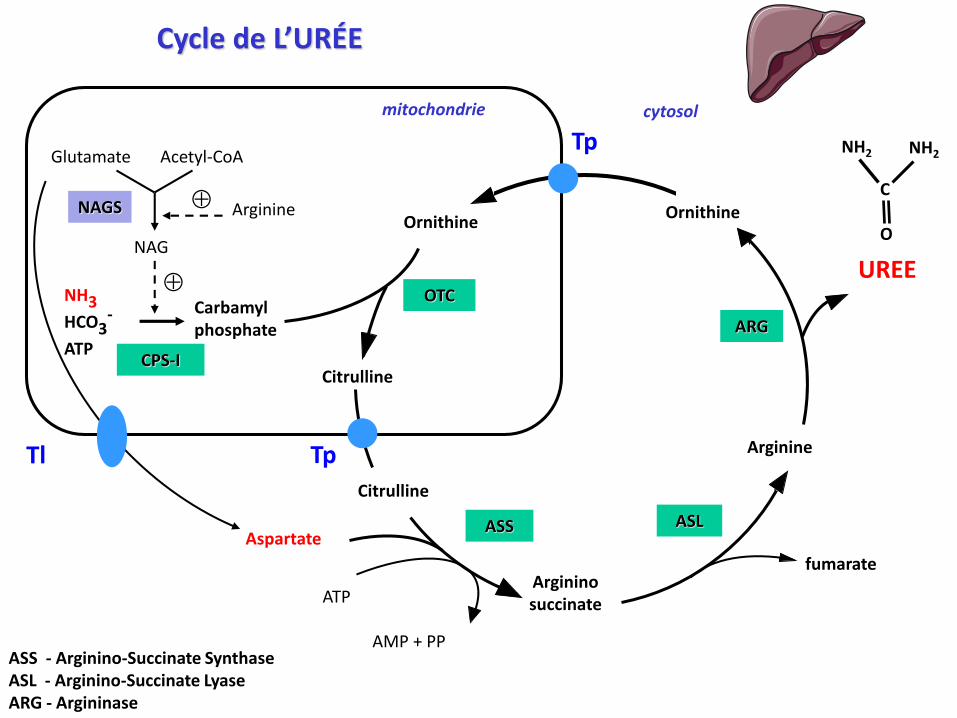
Ornithine
Arginine
Argininosuccinate
Citrulline
Ornithine
Citrulline
Carbamylphosphate
NH3HCO3
-
ATP
NAG
Glutamate Acetyl-CoA
UREE
fumarate
Aspartate
ATP
AMP + PP
Arginine
Cycle de L’URÉE
NAGS
CPS-I
OTC
ASS ASL
ARG
mitochondrie cytosol
Tp
TpTl
C
O
NH2NH2
ASS - Arginino-Succinate SynthaseASL - Arginino-Succinate LyaseARG - Argininase
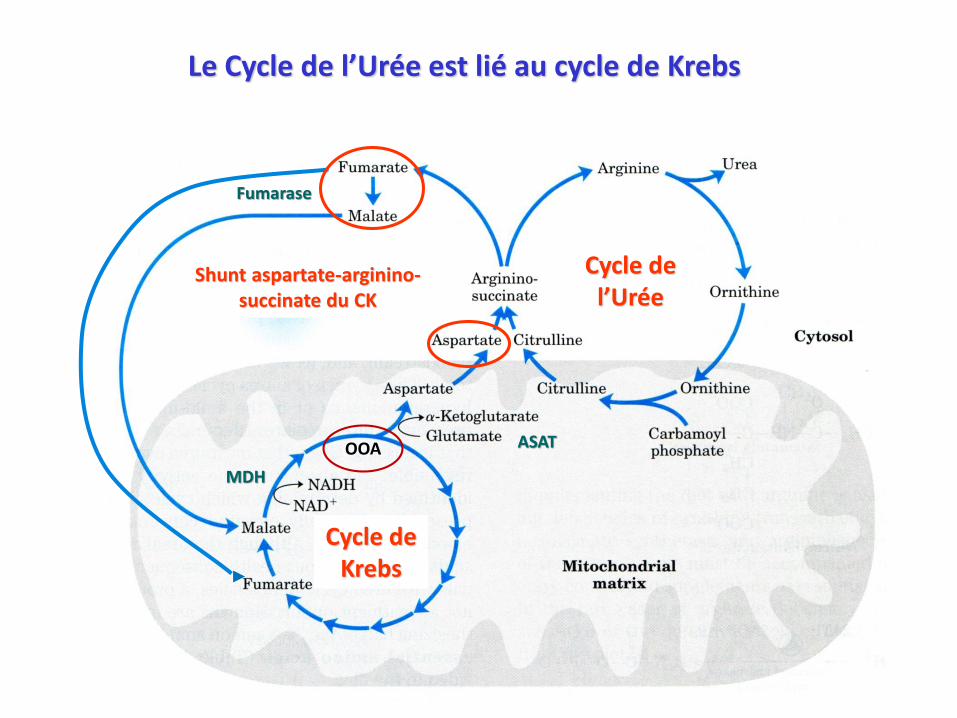
OOA
Le Cycle de l’Urée est lié au cycle de Krebs
Cycle deKrebs
Cycle del’Urée
Shunt aspartate-arginino-succinate du CK
ASAT
MDH
Fumarase
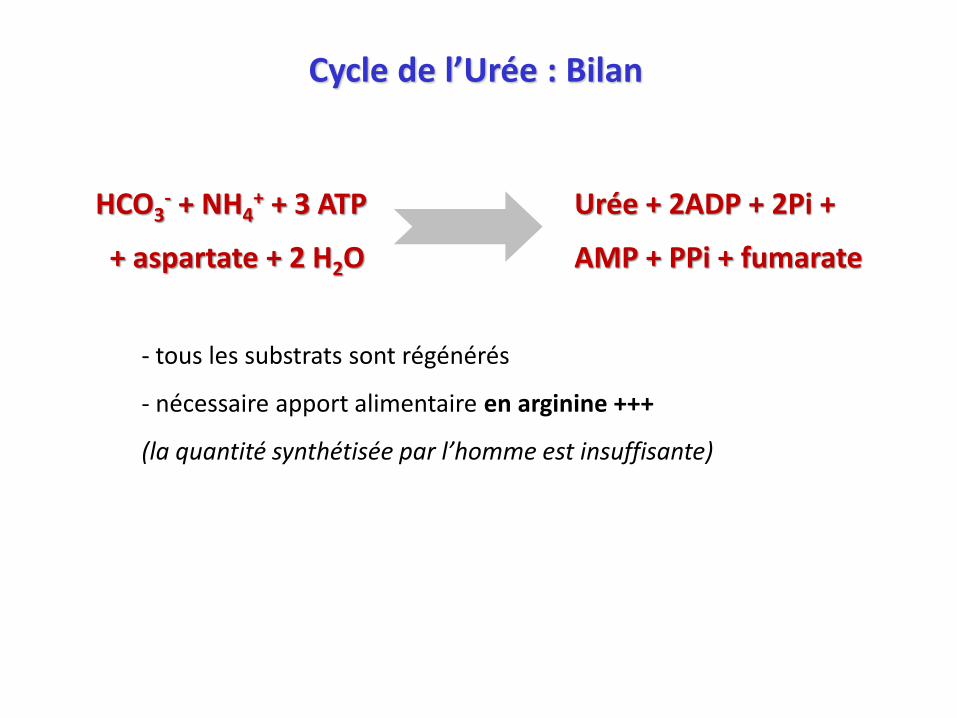
Cycle de l’Urée : Bilan
HCO3- + NH4
+ + 3 ATP Urée + 2ADP + 2Pi +
+ aspartate + 2 H2O AMP + PPi + fumarate
- tous les substrats sont régénérés
- nécessaire apport alimentaire en arginine +++
(la quantité synthétisée par l’homme est insuffisante)

Cycle de l’Urée : REGULATION
3 types de régulation :
Par allostérie
Disponibilité du substrat
Hormonale
Cycle de l’Urée : REGULATION
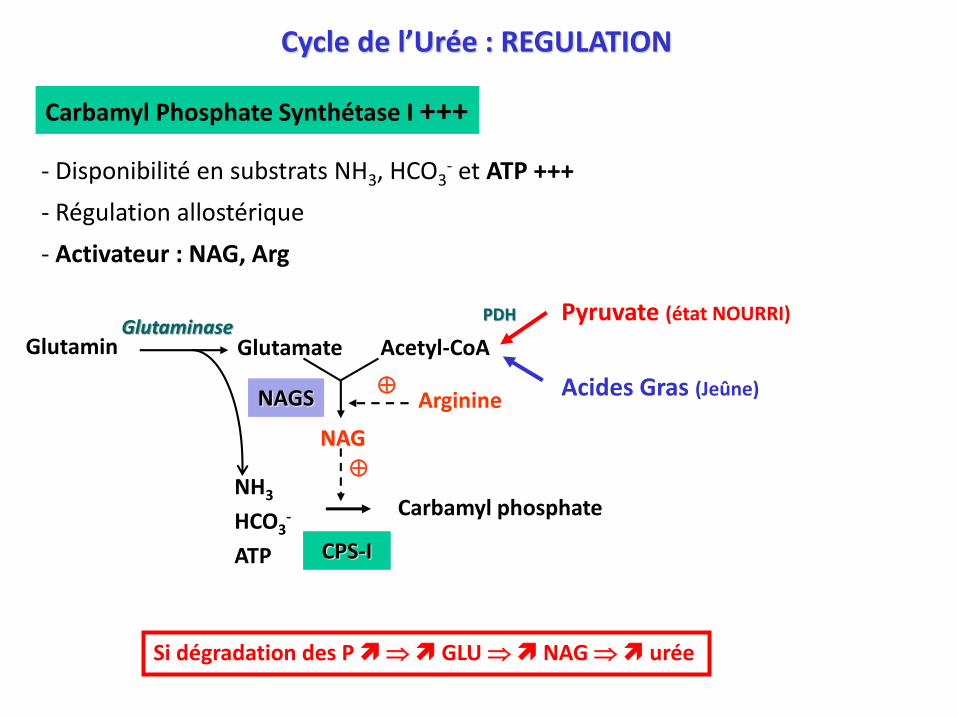
Carbamyl Phosphate Synthétase I +++
- Disponibilité en substrats NH3, HCO3- et ATP +++
- Régulation allostérique
- Activateur : NAG, Arg
Pyruvate (état NOURRI)PDH
Acides Gras (Jeûne)
Si dégradation des P GLU NAG urée
Carbamyl phosphateNH3
HCO3-
ATP
NAG
Glutamate Acetyl-CoA
Arginine
NAGS
CPS-I
GlutaminGlutaminase
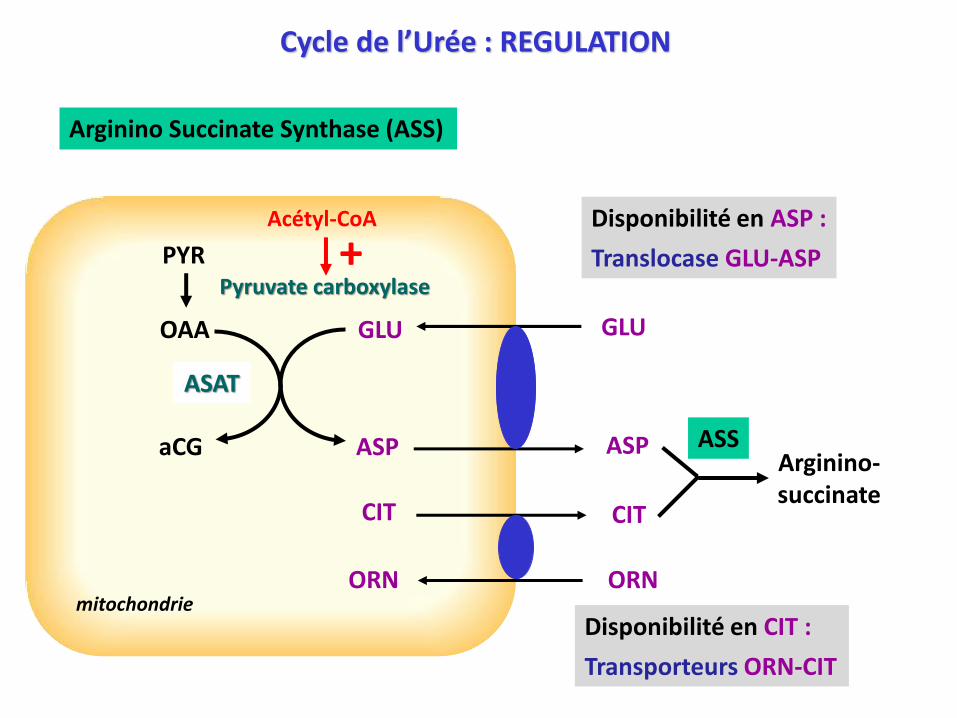
Arginino Succinate Synthase (ASS)
Disponibilité en ASP :
Translocase GLU-ASPPYRPyruvate carboxylase
Acétyl-CoA
+
ASPArginino-succinate
ASS
OAA
aCG
GLU
ASP
ASAT
GLU
CIT
ORN
CIT
ORNmitochondrie
Disponibilité en CIT :
Transporteurs ORN-CIT
Cycle de l’Urée : REGULATION
Cycle de l’Urée : REGULATION
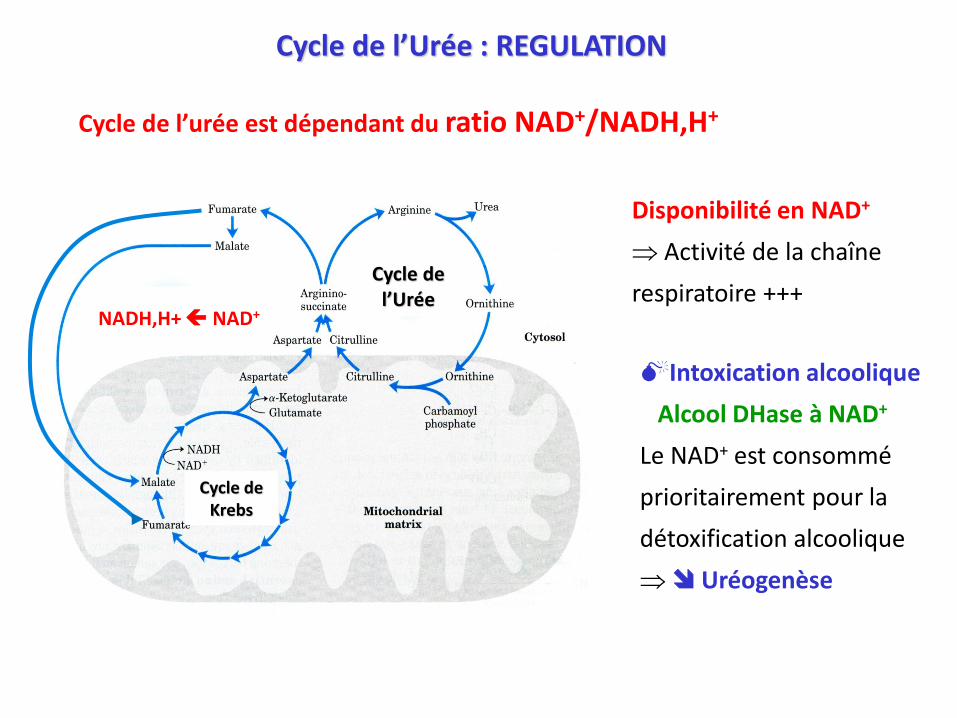
Cycle de l’urée est dépendant du ratio NAD+/NADH,H+
Disponibilité en NAD+
Activité de la chaîne
respiratoire +++
Intoxication alcoolique
Alcool DHase à NAD+
Le NAD+ est consommé
prioritairement pour la
détoxification alcoolique
Uréogenèse
Cycle deKrebs
Cycle del’Urée
NADH,H+ NAD+

Cycle de l’Urée : Régulation au cours de différent états nutritionnels
Période post prandiale
- 50% de l’azote absorbé est transformé en urée
Au niveau intestinal :- glutamine et glutamate totalement oxydés dans l’entérocyte- autres AA sont absorbés et passent dans la circulation portale- flux d’AA et d’ammoniac vers le foie
Au niveau hépatique :- Gln = Glu + NH3 par la glutaminase des hépatocytesavec une activité très élevée car l’enzyme est activée par le NH3.- alanine transformée en aspartate qui peut rentrer dans le cycle de l’urée

Période de jeûne modéré (1-3 jours)- le processus d’épargne protéique consomme les substrats uréogéniques- AA- néoglucogenèse
Période de jeûne prolongé (>3 jours)- la protéolyse (bilan azoté négatif) stimule la synthèse de l’urée
Apports exogènes : Régimes hyperprotidiques
Apports endogènes : Etats d’hypercatabolisme protéique
- Brûlés
- Traumatismes
Cycle de l’Urée : Régulation au cours de différent états nutritionnels

Cycle de l’Urée: REGULATION HORMONALE
Cortisol : catabolisant ; augmente la protéolyse et
l’efflux musculaire et donc augmente l’uréogenèse
Glucagon : catabolisant; augmente le transport
hépatocytaire des AA et donc augmente l’uréogenèse
Insuline : anabolisante (muscle+++) ; oriente les AA vers
la synthèse protéique et diminue l’uréogenèse
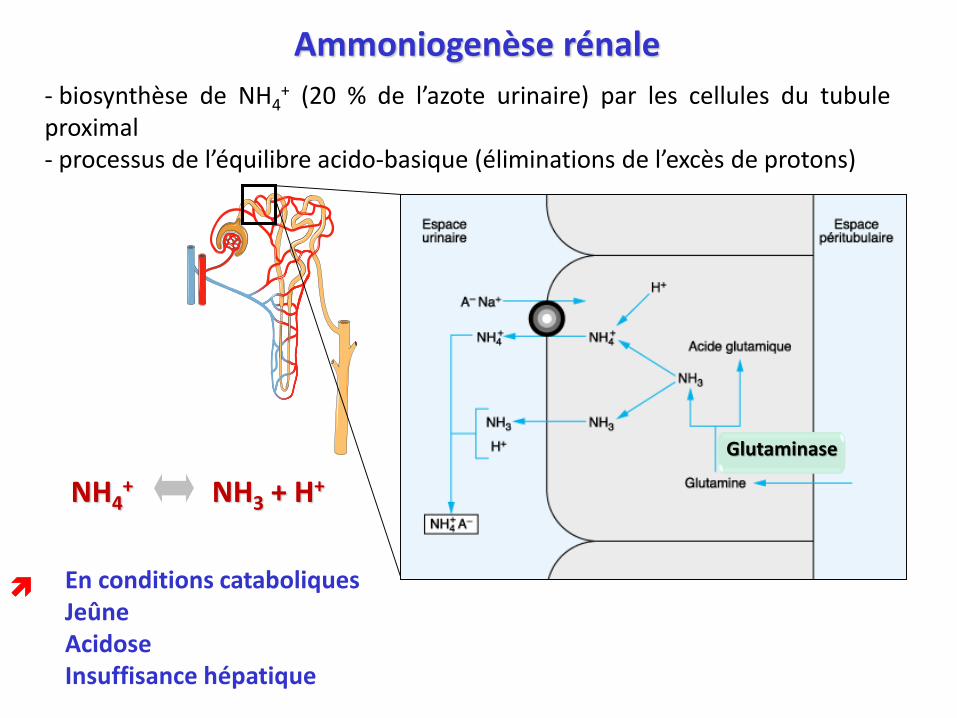
Ammoniogenèse rénale
- biosynthèse de NH4+ (20 % de l’azote urinaire) par les cellules du tubule
proximal- processus de l’équilibre acido-basique (éliminations de l’excès de protons)
En conditions cataboliquesJeûneAcidoseInsuffisance hépatique
NH4+ NH3 + H+
Glutaminase

PATHOLOGIE
Hyperammoniémie :élévation de la concentration de NH4
+ dans le sangadulte > 100 μmol/L (zone grise 50-100 μmol/L)
Etiologie des hyperammoniémies :
- primaires - héréditaires
- secondaires
Les valeurs normales de l’ammoniémie :
14 - 55 μmol/L 11 - 48 μmol/L
nouveau-né - hyperammoniémie transitoire > 100 μmol/L

Manifestations cliniques
Encéphalopathie œdème cérébral
Atteinte digestive et hépatique
Troubles psychiatriques
aucune spécificité !
Tableau aigu ou chronique
A tout âge - période néonatale âge adulte

Hyperammoniémies secondaires
Insuffisance Hépatique sévère +++
- Causes : hépatites aiguës (virales, toxiques…), cirrhose
Acidose défaut d’élimination urinaire (NH4+); insuffisance rénale.
Anomalies héréditaires du métabolisme :
- Acidurie organique (+ acidose métabolique)
- Déficit b-oxydation des AG
- Déficit chaîne respiratoire
Médicaments (valproate)
( urée)
Prématurité : immaturité hépatique & défaut de perfusion

Hyperammoniémies PRIMAIRES
Déficits en Enzymes du cycle de l’urée
OMIM≠ (Online Mendelian Inheritance in Man) Transmission Prévalence
237310 - NAGS
237300 - CPS I
311250 - OCT
215700 - ASS
207900 - ASL
207800 - Arginase
Ar
Ar
Lié à l’X
Ar
Ar
Ar
1/ 62000
1/ 14000
1/ 57000
1/ 70000
1/ 363000
www.ncbi.nlm.nih.gov/omim
Maladies héréditaires du métabolisme de l’urée
Déficits des Transporteurs
238970 - ORNT1 (SLC25A15 ORN/CIT)
603471 - CITRINE (SLC25A13 GLU/ASP)
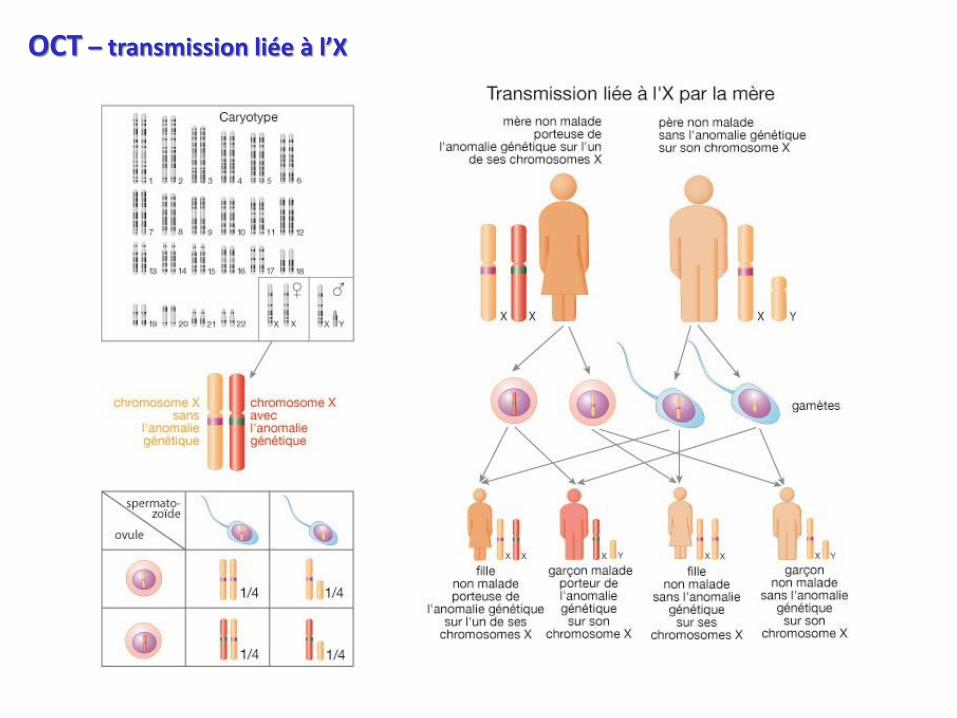
OCT – transmission liée à l’X
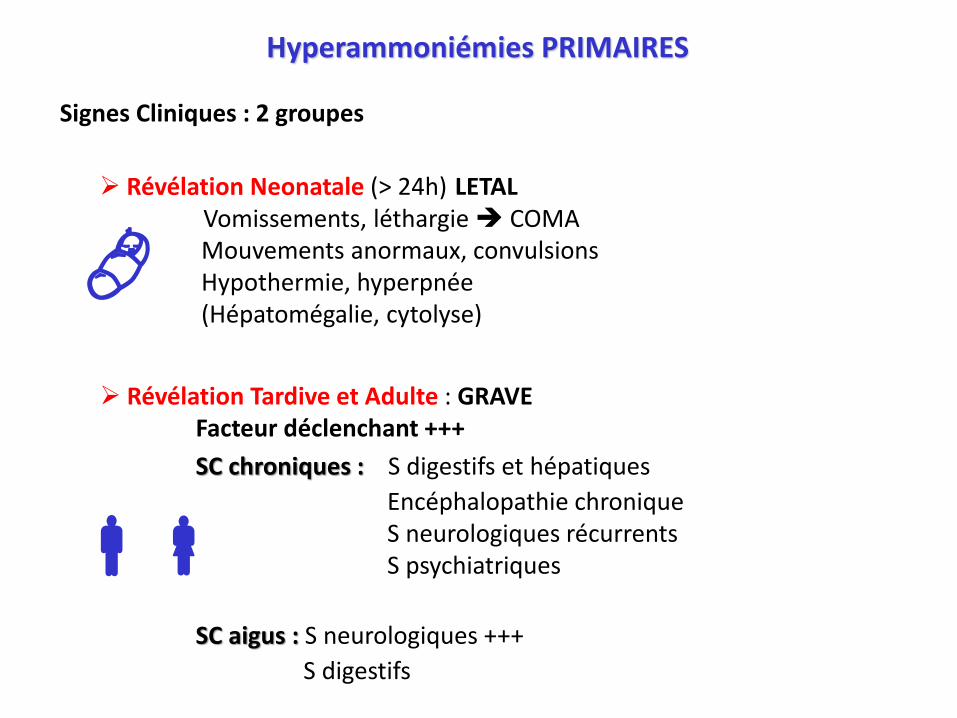
Révélation Neonatale (> 24h) LETALVomissements, léthargie COMAMouvements anormaux, convulsionsHypothermie, hyperpnée(Hépatomégalie, cytolyse)
Révélation Tardive et Adulte : GRAVEFacteur déclenchant +++
SC chroniques : S digestifs et hépatiques
Encéphalopathie chroniqueS neurologiques récurrentsS psychiatriques
SC aigus : S neurologiques +++
S digestifs
Hyperammoniémies PRIMAIRES
Signes Cliniques : 2 groupes

Ammoniémie plasmatique
Conditions pré-analytiques +++
- prélèvement sur anticoagulants (héparinate, EDTA)
- acheminée rapidement au laboratoire <15min
- dans la glace
(à T°Ambiante GLN GLU + NH3 – surestimation!!!)
- interférence analytique – hémolyse
- centrifugation du tube et analyse – le plus rapidement possible
(plasma congelé)
Cycle d’ammoniémie : dosages répétés au cours de la journée avant et
après les repas (formes à révélation tardive - élévation progressive
au cours de la journée)
Hyperammoniémie - outil diagnostique biologique

- Chromatographie des AA plasmatiques et urinaires- transporteurs du NH3 – glutamine, proline, glycine, alanine- AA du cycle de l’urée
- Chromatographie des Ac organiques urinaires (CAO)- Diagnostic des aciduries organiques- Orientation vers un déficit de la b-oxydation- Identification de l’acide orotique et de l’uracile
- Acide orotique urinaire (Carbamylphosphate Ac orotique Urines)
- Base pyrimidique synthétisée dans le cytosol- Synthèse à partir du CP accumulé dans la mitochondrie- Diagnostic différentiel des déficits en aval de la synthèse du CP
(NAGS, CPS, OTC)
- Biologie Moléculaire
- Dosages enzymatiques spécifique
- CPS, OTC : cellules hépatiques; intestinales- ASL, ASS : fibroblastes- Arginase : érythrocytes
Hyperammoniémies - outils diagnostiques biologiques
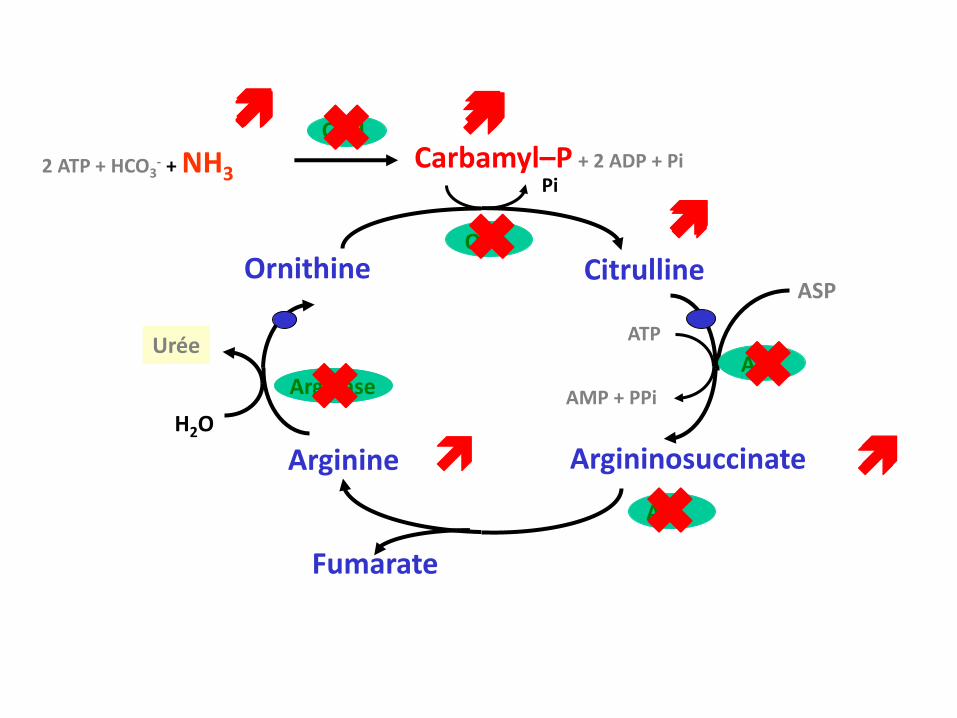
CPSI
2 ATP + HCO3- + NH3
Carbamyl–P + 2 ADP + Pi
Arginine
Fumarate
ASP
Argininosuccinate
ATP
AMP + PPi
ASSArginase
H2O
Urée
Ornithine Citrulline
Pi
OTC
ASL

Apport suffisant en AA essentiels
Apport d’arginine +++
Hyperammoniémies – prise en charge
Elimination des métabolites toxiques- Épuration de l’azote excédentaire : Hémodialyse +++- Épuration par des chélateurs iv : Phénylbutyrate, Benzoate
(stimulent une voie métabolique alterne)
Limitation de la néoformation des métabolites toxiques- Limiter l’apport d’azote : Régime pauvre en protéines- Éviter le catabolisme: Apport calorique suffisant (glucidolipide)
Transplantation hépatique

Diagnostic rapide +++ Prise en charge du patient – urgence vitale!
DPN +++ (villosités choriales, LA)
Les gènes de toutes les enzymes sont connus (cf OMIM)
Mutations décrites pour toutes les maladies
Centres Spécialisés de prise en charge:
« Centre de Référence Maladies Rares (CRMR) »
- Compétences Cliniques
- Compétences Biochimiques
(analyses des métabolites, enzymes, biologie moléculaire)
Hyperammoniémies – prise en charge

Cas cliniques

Mathieu
• 1er enfant de parents non consanguins et sans ATCD
• Grossesse, accouchement : N
• 3 mois et demi : introduction d’un lait artificiel
– Mauvaise prise des biberons – vomissements irréguliers
– Mauvaise prise de poids – Modification du lait
• 5 mois : – Hospitalisation pour vomissements, anorexie et cassure de la courbe
– Diagnostic : reflux gastro-oesophagien
Modification du lait, motilium + polysilane
• 8 mois – Fatigué, refuse de manger – pousse des hurlements, crise généralisée
• Hospitalisation – Inconscient avec mouvements de pédalage
– Débord hépatique (1,5 cm) – Convulsions
– Bilan : alcalose respiratoire; NH3 : 410 μmol/L
• Evolution : décès 48 heures plus tard (œdème cérébral)
Déficit en OCT

François 14 ans
• ATCD – Développement staturo-pondéral normal
– Céphalées intermittentes
– Décès par coma inexpliqué d’un oncle maternel alors âgé de 30 ans
• De J0 à J5 – Céphalées, flou visuel, anorexie, vomissements
– Examen clinique; NFS, ionogramme, transaminases : normaux
• J6 – Hallucinations, confusion
– Hospitalisation en pédiatrie puis en réanimation
• De J7 à J9 – Coma d’aggravation progressive
– Scanner, PL, glycémie, iono, toxico : N
– Ammoniémie : 344 μmol/L puis 755 μmol/L
– Traitement : régime sans protéines, alimentation glucido-lipidique, épurateurs
• J9 : décès
Déficit en OCT

Monsieur J… 49 ans
• Pas d’ATCD familiaux et personnels évocateurs - coureur de semi marathon
• J0 – Vomissements au retour d’un semi marathon
• De J4 à J6 – Asthénie physique suivie d’un épisode de vomissements et de diarrhées
– stupeur, ralentissement de la gestuelle, soif
Crises d’agitation, agressivité verbale, incoordination psychomotrice, obnubilation, désorientation.
Appel du médecin traitant : refus des soins, déambulation
• J7 à 0h00 – Admission aux urgences puis transfert en Neurologie à 12h30
– Obnubilation, somnolence, vomissements, bradypsychie, trouble de l’élocution, fixité du regard,stéréotypie gestuelle, bâillements
– Biologie standard normale, pas de cytolyse hépatique, absence de toxiques, PL normale,
alcalose ventilatoireSuspicion d’état de mal épileptique
• J7 à 22h – Transfert en Réanimation médicale – Glasgow à 5/15, hypertonie pyramidale franche,mydriase bilatérale réactive, œdème cérébral
Encéphalopathie d’étiologie indéterminée
– Hyperammoniémie à 700 μmol/L ! Aggravation rapide
• J10 – Décès du patient – ammoniémie à 1859 μmol/L
Déficit en OCT

1. Accident aigu neuro-hépato-digestif
2. Association ou succession de symptômes hépato-digestifs et neuropsychiatriques de présentation aiguë ou chronique
3. Tableau monosymptomatique qui ne fait pas sa preuve
• vomissements, anorexie, cassure de courbe
• anorexie et atteinte hépatique
• retard psychomoteur, épilepsie
• accès d’agitation, agressivité
• syndrome confusionnel isolé
• coma
Quand doser l’ammoniémie ?